
Cerca i descobriment de selenoproteïnes
L’objectiu del present treball és anotar les selenoproteïnes del genoma del Nannospalax galili, així com la maquinària per a la correcta formació d’aquestes proteïnes.
Com que els gens que codifiquen selenoproteïnes contenen codons UGA, els quals són normalment utilitzats com a senyals stop durant la traducció, no acostumen a estar anotades en les bases de dades de seqüències. Hi ha dues aproximacions bioinformàtiques que han resultat molt útils per a la identificació de selenoproteïnes.
La primera aproximació es basa en el fet que cada gen de selenoproteïnes posseeix un codó UGA en la pauta de lectura que codifica per Sec i també un element SECIS. La sensibilitat d’aquesta prova va millorar amb l’anàlisi comparatiu d’elements SECIS corresponents a selenoproteïnes ortòlogues de genomes d’espècies estretament relacionades.
La segona aproximació consisteix en identificar selenoproteïnes mitjançant la cerca de codons UGA a la pauta de lectura mitjançant anàlisis de seqüències adjacents al UGA en genomes completament seqüenciats. Com que gairebé totes les selenoproteïnes contenen homòlegs en els quals Sec és reemplaçada per Cys, aquest mètode va permetre la identificació de selenoproteïnes independentment dels elements SECIS (que presenten variació entre espècies). Aquesta aproximació també es pot utilitzar per la identificació de residus Cys catalítics amb funció redox en enzims que no siguin selenoproteïnes.
Actualment, l’eina que s’utilitza per a la cerca de selenoproteïnes en eucariotes és el Seblastian. Aquesta, combina ambdues aproximacions anteriors. La recerca de les selenoproteïnes de N. galili s’ha realitzat a partir de la comparació amb els genomes d’Homo sapiens i d’Echinops telfairi que ja es trobaven anotats. A continuació es detalla el procediment.
Obtenció de les querys
En primer lloc, s’han seleccionat aquelles selenoproteïnes i proteïnes de la maquinària de síntesi de les mateixes que volem comparar amb el genoma de Nannospalax galili. S’han escollit les proteïnes d’Homo sapiens i d’Echinops telfairi presents a SelenoDB. Es tracta d’una base de dades que forma part d’un projecte col·laboratiu de llarg termini. Proporciona un marc fiable (base de dades i interfície) per al recull de les anotacions, tant experimentals com computacionals, d’alta qualitat de selenoproteïnes individuals i selenoproteomes.
El genoma d’Echinops telfairi és el genoma més proper que es troba en aquesta base de dades i, és per això, que l’hem utilitzat per a estudiar les selenoproteïnes del nostre genoma d’interès. Tanmateix, aquest organisme no té correctament anotades totes les proteïnes i, per aquest motiu, també s’han obtingut les seqüències d’aminoàcids (querys) del genoma d’Homo sapiens ja que és el que està anotat de manera més acurada. És important destacar que aquelles querys que contenien l’aminoàcid selenocisteïna (abreviat amb una U) s’han hagut de modificar. S’han hagut d’intercanviar les U per X, ja que els programes executats no reconeixen la U com a caràcter vàlid.
Obtenció del genoma de Nannospalax galili
Els responsables de l’assignatura ens han facilitat el genoma de Nannospalax galili a partir del qual s’ha realitzat aquest treball. Per a poder extreure’l cal introduir la següent comanda al terminal:
Cerca de selenoproteïnes
Abans d’iniciar qualsevol cerca de selenoproteïnes en el genoma d’estudi, ha estat necessari exportar els diferents programes amb les comandes següents:
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH # pel NCBI Blast
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/ # pel NCBI Blast
$ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH # per l'exonerate
$ export PATH=/cursos/BI/soft/t_coffee/i386/bin:$PATH # pel t_coffee
Cerca automatitzada de selenoproteïnes
Per tal d’agilitzar el procés de cerca de selenoproteïnes en el genoma de Nannospalax galili, s’ha realitzat el següent programa en llenguatge Perl. Quan el programa s’executa, analitza si la query donada es troba en el genoma d’interès o no.
Per a que funcioni correctament, cal que el programa estigui al luke, el clúster utilitzat per les assignatures de Bioinformàtica i Biologia Estructural dels estudis de Biologia Humana i del Màster en Bioinformàtica per a les Ciències de la Salut, a la mateixa carpeta que la query.
Cerca manual de selenoproteïnes
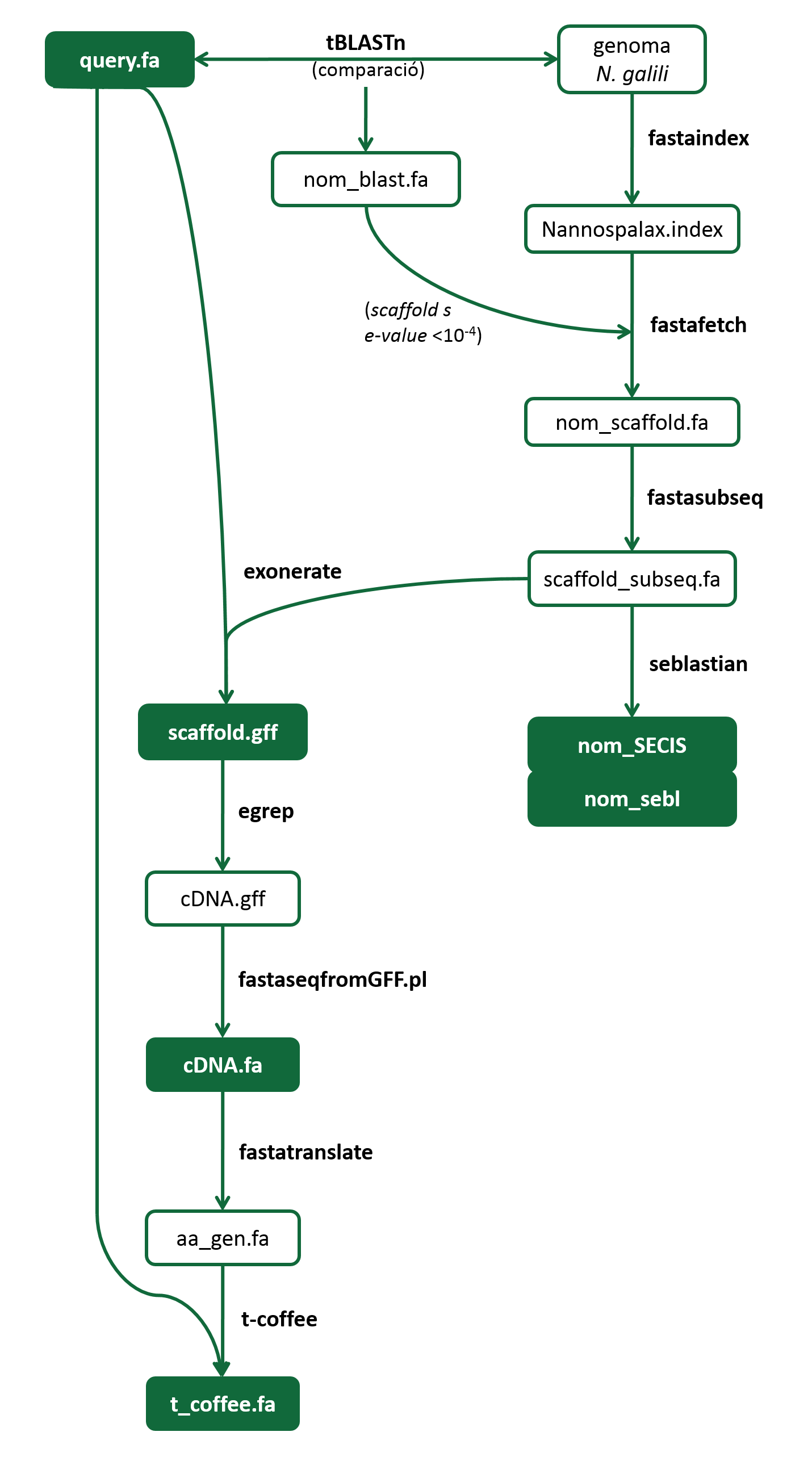
Per a conèixer millor quins han estat els programes utilitzats i les funcions de cadascun d’aquests, seguidament s’exposa com es realitza la cerca manual de selenoproteïnes mitjançant un esquema i les instruccions específiques per a cada pas.
Alineament de seqüències en BLAST
Primerament, cal identificar quines són les regions del genoma de Nannospalax galili que són susceptibles a contenir selenoproteïnes. Per aquest motiu, es fa un BLAST de la query contra el genoma d’interès.
El BLAST (Basic Local Alignement Search Tool) és un programa que permet fer alineaments locals d’una seqüència problema (query) amb seqüències provinents d’una bases de dades, en aquest cas el genoma de N. galili. El programa segueix un algorisme heurístic a partir del qual selecciona aquelles seqüències o hits amb una homologia més gran en referència a la query.
En el nostre cas concret, hem utilitzat el tBLASTn ja que és capaç de comparar una seqüència d’aminoàcids (query) amb una base de dades de nucleòtids (genoma). Aquesta variació del BLAST tradueix totes les seqüències de nucleòtids en els 6 ORFs (Open Reading Frames) possibles, les compara amb la seqüència problema i, finalment, et mostra aquells hits amb major homologia. La comanda utilitzada per realitzar aquesta tasca ha estat:
On:
- -p vol dir quin tipus de BLAST s’utilitza, en aquest cas tblastn
- -i és la seqüència problema o query
- -d indica la base de dades
- -o és el fitxar de sortida del tBLASTn
- -m8 és el format amb el qual volem que aquest fitxer de sortida estigui estructurat
El fitxer de sortida del tBLASTn conté una llista dels scaffolds o regions del genoma que tenen una alta similitud amb la query. Per a poder valorar la significança de cada scaffold cal tenir el compte el seu valor de e-value, que descriu el nombre de seqüències que poden tenir similitud amb la seqüència problema només per atzar. Com més petit és el e-value d’un scaffold més elevada serà la seva homologia. Nosaltres hem considerat que un e-value és significatiu quan és igual o inferior a 10-4.
Extracció de la regió genòmica d’interès
Per a poder continuar l’estudi, cal extreure del genoma de Nannospalax galili la regió genòmica amb la qual el tBLASTn troba similitud, és a dir, aquelles seqüències que podrien codificar per una selenoproteïna.
En primer lloc, cal fer un índex del genoma de N.galili a partir de les següents comandes:
On:
- /cursos/BI/genomes/vertebrates/2014/Nannospalax_galili/genome.fa indica la ubicació del genoma
- Nannospalax.index és el fitxer de sortida
Un cop obtingut l’índex s’han d’extreure aquells scaffolds que han obtingut un e-value significatiu en realitzar el BLAST. És per això que s’introdueix al terminal la següent comanda:
On:
- /cursos/BI/genomes/vertebrates/2014/Nannospalax_galili/genome.fa indica la ubicació del genoma
- “nom_scaffold” és el nom de la regió que volem extreure
- nom_scaffold.fa és el nom del fitxer de sortida que conté la regió extreta
Com que es vol extreure el gen sencer que conté la proteïna buscada, es selecciona la regió que envolta el gen per tal d’assegurar-se que s’agafa l’element SECIS corresponent. És per això que s'ha de seleccionar una posició d'inici i una de final, per a fer-ho s'inicia, per exemple, 15.000 nucleòtids abans del primer nucleòtid alineat amb el BLAST i s'agafa una llargada de 30.000 parells de bases. Aquests valors poden ser modificats per cada proteïna:
On:
- /cursos/BI/genomes/vertebrates/2014/Nannospalax_galili/genome.fa indica la ubicació del genoma
- start és la posició del primer nucleòtid de la seqüència de N.galili que s’alinea amb el BLAST - 15.000
- length és 30.000
- scaffold_subseq.fa és el fitxer de sortida
Anotació genòmica
Exonerate
L’Exonerate és un programa que prediu la presència de gens en una seqüència. Permet obtenir un bon alineament i predicció acurada de la composició exònica de la regió. Per aquest motiu, s’alinea la seqüència extreta amb el fastasubseq amb la seqüència de DNA de la query inicial seguint la comanda següents:
On:
- -m p2g és el model d’alineament, en el nostre cas és una query contra la seqüència d’un genoma
- showtargetgff ens mostra el fitxer de sortida en format GFF
- exhaustive fa una predicció exhaustiva, mostrant fins i tot alineaments subòptims
- -q ubicació de la query
- -t ubicació del resultat del fastasubseq
- scaffold.gff és el fitxer de sortida
Si a Nannospalax galili es conserva la selenocisteïna, al fitxer de sortida en format GFF hauríem de trobar la X de la query (recordem que correspon a la U) alineada amb asteriscs (*) ja que el programa ho interpreta com un codó stop. Per extreure les seqüències exòniques del fitxer anterior cal executar la següent comanda:
On:
- -w és el patró que volem extreure, en el nostre cas són els exons
- scaffold.gff de quin arxiu ho volem extreure
- cDNA.gff és el fitxer de sortida on s’introdueixen els resultats
Un cop obtinguda la informació de quina és la seqüència d’aminoàcids dels exons cal aplicar-ho al fitxer provinent del fastasubseq per a poder extreure la seqüència nucleotídica d’aquests:
On:
- scaffold_subseq.gff és el fitxer de sortida del fastasubseq
- cDNA.gff és el fitxer que conté el exons
- cDNA.fa és el fitxer que contindrà la seqüència de nucleòtids dels exons
A continuació, s’ha de traduir la seqüència de bases nitrogenades obtinguda en el pas anterior per a poder conèixer quina és la proteïna que codifiquen. El fastatranslate proporciona un fitxer de sortida amb els sis marcs de lectura possibles (ORFs). Tot i això, nosaltres només seleccionem el primer marc de lectura ja que és el que amb una probabilitat molt alta serà el correcte:
On:
- -F 1 indica que només ha de llegir el primer ORF
- cDNA.fa és el fitxer a traduir
- aa_gen.fa és el fitxer de sortida que conté els exons traduïts
T-coffee
El T-Coffee (Tree-based Consistentcy Objective Function for alignment Evaluation) és un mètode per a l’alineament de seqüències múltiples. Es tracta d’un alineament progressiu, és a dir, considera informació de totes les seqüències en cada pas de l’alineament i no només aquelles que s’estan alineant. Amb aquest programa podrem alinear el resultat de la traducció anterior amb la seqüència de la query de la que es parteix. El resultat del T-Coffee ens indicarà si hi ha homologia o no entre ambdues seqüències, a més de mostrar l’alineament al terminal. La comanda necessària per a poder executar el programa és:
On:
- query.fa és la proteïna extreta de la base de dades
- aa_gen.fa és el fitxer obtingut del fastatranslate
- t_coffe.fa és el fitxer de sortida d’aquest programa que conté l’alineament entre les dues proteïnes
Cerca d’elements SECIS
Per a finalitzar, s’han de predir els possibles elements SECIS que poden contenir aquestes proteïnes al seu extrem 3’ downstream. Per fer-ho cal utilitzar el programa Seblastian que es troba disponible online. Aquest programa utilitza en primer lloc, l’eina SECISearch3 per trobar tots els elements SECIS potencials predits en una seqüència diana mitjançant el reconeixement de patrons. Aleshores, examina totes les seqüències upstream d’aquests candidats per esbrinar si conserven l’estructura de selenoproteïna. Per a que el programa pugui fer la seva funció cal introduir el fitxer de sortida obtingut a partir del fastasubseq. Hem guardat l’output, tant de l’estructura del SECIS com de la predicció del Seblastian.