 Figura 1
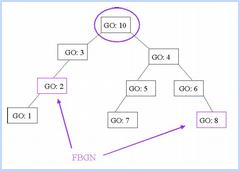
Figura 1Para realizar este proyecto hemos utilizado Drosophila melanogaster como organismo modelo. En primer lugar, obtuvimos la anotación de su genoma, en la base de datos Gene Ontology, la cual describe como se comportan los productos de los genes en el contexto celular.
El archivo que nos descargamos es gene_association.fb. Sobre este archivo, los campos que nos interesan son: GO:XXXXXXX y FBgn:ZZZZZZZ. Para esto los comandos utilizados fueron:
$ cut -f 2 > GO.txt
$ cut -f 5 > FB.txt
$ paste GO.txt FB.txt > anotacion.txt
Obtuvimos una tabla del tipo:
| GO:XXXXXXX | FBgn:ZZZZZZZ |
| GO:0044523 | FBgn:0127452 |
| .......... | ............ |
En anotacion.txt tenemos asociados cada gen (FBgn) a una función (GO). A continuación queremos obtener los ancestros de cada término GO. Para ello nos descargamos el archivo go_expanded_filtered.txt y utilizamos el programa1.pl sobre éste.
El programa va abrir también el archivo ordenado.txt, obtenido a partir de ejecutar:
$ anotacion.txt | sed 's/GO://' | sort +0n > sinGO.txt
$ sinGO.txt | sed 's/^/GO:/' > ordenado.txt
De esta forma, al estar ordenados los GO numéricamente, al programa le sera más fácil encontrar el mismo término GO en el archivo de los ancestros.
Por lo tanto, en el GO_VS_FBgn_s.txt (output del programa) tenemos el término GO inicial más sus ancestros. Como un mismo gen puede tener distintas funciones, va a encontrarse repetido en términos superiores (Fig.1).
Figura 1Para eliminar los términos repetidos utilizaremos los siguientes comandos:
$ sort GO_VS_FBgn_s.txt | uniq > sinrepeticion1.txt
Posteriormente se ejecuta el programa2.pl sobre sinrepeticion1.txt para obtener por cada término GO todos los genes anotados, pertenecientes a esa categoria. Así el output (tabla.txt) debe tener este formato:
GO:0001413 FBgn001014 FBgn001511 FBgn011552
GO:0000711 FBgn000268 FBgn008415
...
Para seleccionar aquellas funciones suficientemente representativas, escogemos las que contengan entre 75 y 125 genes. Para ello aplicamos el comando:
$ gawk ' {if ((NF > 76 && (NF < 126 )) print $0, NF -1}' tabla.txt > tablaseleccionados.txt
Por último, para ver si estos genes (Fbgns) que hemos agrupado bajo la misma función (término GO) se encuentran agrupados formando clusters en el genoma, utilizamos el programa cluster_genomic2_allPerl.pl. A este programa le introducimos tantos archivos como términos GO hemos seleccionado, y además un único archivo con todos los genes de Drosophila Melanogaster.
Para crear cada uno de los archivos aplicamos el programa ,multiplesGO.pl sobre el archivo tablaseleccionados.txt, de forma que cree en la carpeta sets todos los archivos que tienen como nombre su término GO. Cada uno contiene todos los genes con esa función, con el término Fbgn.
Pero para que el programa cluster_genomic2_allPerl.pl funcione hemos sustituido el identificador de los genes de la base de datos FlyBase (FBgn) por el que tienen en la base de datos RefSeq. Esta transición la hacemos mediante la tabla FBgn_Sym_CG_4-0.txt, que contiene las equivalencias, y el script synonyms.pl, de la siguiente forma:
$ ls sets | while read file; do perl synonyms.pl -- list sets/$file -- fb FBgn_Sym_CG_4-0.txt -- rl refLink.txt -- out r | grep NM > refseqs/$file; done
Refseqs es la carpeta a la que hemos redireccionado cada uno de los archivos.
Hemos creado el archivo todos_genes_fbgns.txt, que como su nombre indica contiene todos los genes del genoma de la mosca, mediante el siguiente comando:
$ sort FB.txt | uniq > todos_genes_fbgns.txt
Como en el caso anterior, transformamos el nombre de los genes de este modo:
$ perl synonyms.pl -- list todos_genes_fbgns.txt -- fb Fbgn_Sym_CG_4-0.txt -- rl refLink.txt -- out r | grep NM > todosNM.txt; done
Finalmente ejecutamos el programa cluster_genomic2_allPerl.pl:
$ ls refseqs/ | while read file; do perl cluster_genomic2_allPerl.pl -- reg refseqs/$file -- ref todosNM.txt -- gwindow 7 -- min_genes 3 > resultados/$file; done
Nota: gwindow hace referencia al tamaño de la ventana utilizado para mapear el genoma, que es de 7 genes; la opción min_genes corresponde al mínimo número de genes que nosotras queremos considerar como un cluster.