ANÀLISI DE LA SEQÜÈNCIA GENÒMICA ANÒNIMA ENr131
- ABSTRACT
L'abril del 2003 va finalitzar la seqüenciació del genoma humà. Des de llavors s'ha posat en marxa el projecte ENCODE (Encyclopedia of DNA elements), projecte dirigit a l'identificació de seqüències funcionals, ja siguin codificants (gens) com reguladores (elements cis). L'objectiu d'aquest treball serà l'estudi d'una seqüència anònima humana pertanyent a aquest projecte mitjançant eines computacionals.
La seqüència a estudi és la ENr131 o NT_005120.14, de 500Kb i localitzada al braç llarg del cromosoma 2 (2q37) (Fig.1).

Fig.1 Localització de la seqüència NT_005120.14 en el cromosoma 2.
La seqüència ha estat descarregada de la base de dades EMBL. Primer s'han localitzat els elements repetitius (SINEs, LINEs...) i després han estat emmascarats mitjançant el programa Repeat Masker, per tal d'analitzar les regions codificants. La predicció de gens 'ab initio' s'ha realitzat mitjançant els programes GENEID, GENSCAN i FGENESH. Posteriorment s'ha validat la predicció utilitzant el programa BLASTN per tal d'alinear la seqüència contra bases de dades de ESTs humans. Comparant els ESTs humans trobats i les prediccions fetes s'han escollit els gens que presentaven major suport i utilitzant el programa BLASTP s'han trobat les proteïnes codificades per aquests gens.
OBTENCIÓ DE LA SEQÜÈNCIA
La seqüència NT_005120.14 ha estat descarregada, en format fasta, de la base de dades de UCSC Genome Browser .
A partir d'aquest document obtenim:
- La llargada de la seqüència, obtinguda utilitzant les següents comandes:
- Posem la seqüència en una sola línia en un nou arxiu:
- $ awk '{printf $1}' sequencia.fa > sequencia.tbl
- Afegim una tabulació entre el nom i la seqüència (en emacs)
- Obtenim la llargada de la 'segona paraula' ($2):
- $ awk '{print length($2)}' sequencia.tbl
- La llargada és: 500000 pb
- El contingut de G+C:
- Tallem la segona paraula en una lletra per línia, les ordenem per odre alfabètic, les comptem i n'obtenim la proporció dividint pel nombre total de pb:
- $ awk '{print $2}' sequencia.tbl | fold -1 | sort | uniq -c | gawk '{print $2,$1/500000}'
- La proporció és dels 4 nucleòtids és de:
- A 0.2692
- C 0.2105
- G 0.216692
- T 0.303608
- I el contingut G+C és del 42.72 %
Informació addicional d'aquest fragment de seqüència es troba en la pàgina de Ensembl i clickar sobre la seqüència ENr131.
ESTUDI DE LA SEQÜÈNCIA
PREDICCIÓ DE GENS 'AB INITIO'
El primer que fem és identificar els elements repetitius que apareixen en la nostra seqüència (NT_005120.14), ja que pertanyen a regions no codificants. Utilitzem el programa Repeat Masker, que es troba en la següent pàgina web: EMBL RepeatMasker server . Els elements repetitius apareixen emmascarats en la seqüència amb la lletra 'N'.
Un cop corregut el programa obtenim els següents arxius:
- repeat.seq.masked, que correspon a la seqüència emmascarada en format fasta.
- repeat.seq.out, que correspon a un llistat de les característiques dels elements repetitius en la seqüència.
- repeat.seq.tbl (Taula 1), que mostra el percentatge d'elements repetitius en la seqüència classificats segons el tipus.
La Taula 1 mostra el nombre de bases codificades per Repeat Masker, un total de 235604, corresponent al 47.12 % de les bases totals de la seqüència (50 kb). El contingut G+C és del 42.12 % (que coincideix amb el calculat amb les comandes anteriors). El tipus de repeticions més abundants són LINEs (23.33 %), seguit de LTRs (9.82 %) i SINEs (6.84 %).
==================================================
file name: repeat.seq.tbl
sequences: 1
total length: 500000 bp (500000 bp excl N-runs)
GC level: 42.72 %
bases masked: 235604 bp ( 47.12 %)
==================================================
number of length percentage
elements* occupied of sequence
--------------------------------------------------
SINEs: 147 34188 bp 6.84 %
ALUs 101 27297 bp 5.46 %
MIRs 46 6891 bp 1.38 %
LINEs: 170 116665 bp 23.33 %
LINE1 120 103823 bp 20.76 %
LINE2 44 12132 bp 2.43 %
L3/CR1 6 710 bp 0.14 %
LTR elements: 101 49117 bp 9.82 %
MaLRs 56 27192 bp 5.44 %
ERVL 21 7306 bp 1.46 %
ERV_classI 23 14128 bp 2.83 %
ERV_classII 1 491 bp 0.10 %
DNA elements: 75 28616 bp 5.72 %
MER1_type 37 7690 bp 1.54 %
MER2_type 12 13132 bp 2.63 %
Unclassified: 1 1711 bp 0.34 %
Total interspersed repeats: 230297 bp 46.06 %
Small RNA: 0 0 bp 0.00 %
Satellites: 0 0 bp 0.00 %
Simple repeats: 56 3392 bp 0.68 %
Low complexity: 40 1959 bp 0.39 %
==================================================
Taula 1 Llargada de la seqüència, percentatge de seqüència emmascarada, proporció G+C, tipus i proporció d'elements repetitius.
A partir de l'arxiu repeat.seq.out, convertit a format gff mitjançant el programa gff2ps, s'ha generat un gràfic (Fig. 2) per visualitzar la distribució dels diferents elements repetitius a la seqüència.
Utilitzem les següents comandes:
- $ grep NT_005120.14 repeat.seq.out | awk 'BEGIN{ OFS="\t" }{ print $5, $11, "repeat", $6, $7, ".", ".", "."; } ' > repeat.seq.out.gff
Correm aquest programa seguint les següents comandes:
- $ gff2ps repeat.seq.out.gff > repeat.seq.out.ps
I visualitzem:
- $ convert -antialias -rotate 90 repeat.seq.out.ps repeat.seq.out.png
- $ kview repepat.seq.out.png
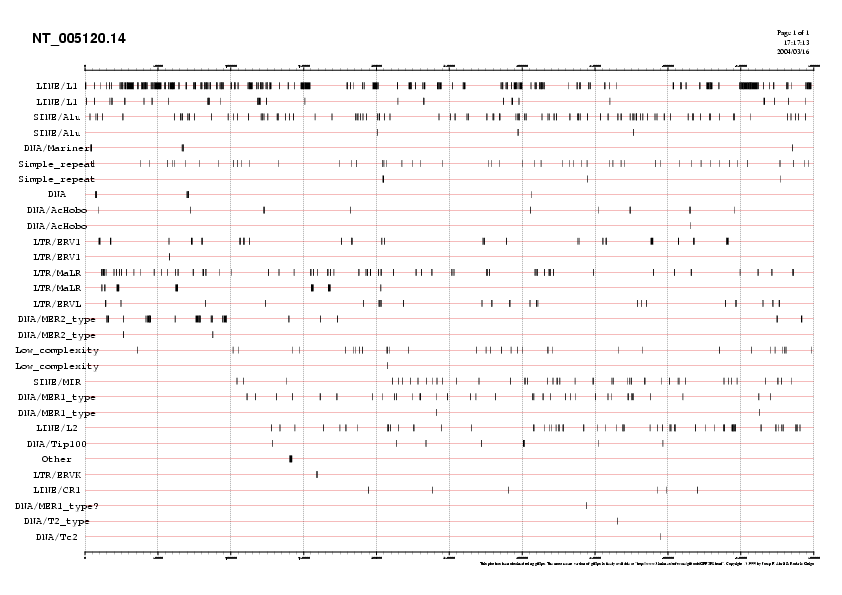
Fig.2 Distribució dels elements repetitius a la seqüència NT_005120.14.
A partir de la seqüència emmascarada en format fasta (repeat.seq.masked) s'han obitngut les prediccions de gens 'ab initio' mitjançant 3 programes de predicció de gens: GENEID, GENSCAN, FGENESH.
Obtenim diferents arxius de text mostrant els gens putatius en la seqüència:
Els arxius s'han manipulat per tal de ser comparables entre ells. La predicció feta per GENEID crea un arxiu de sortida en format gff. En el cas dels altres programes aconseguim arxius de sortida txt. Un cop passats a format gff, els 3 arxius es visualitzen en forma de gràfics.
Fem servir les següents comandes:
Per GENSCAN:
- $ gawk 'BEGIN{OFS="\t"} $2 ~ /Term|Intr|Init/ {print "NT_005120.14", "genscan", $2, start=($4<$5 ? $4 : $5), end=($5<$4 ? $4 : $5), $13, $3, $7, $1;}' NT_005120.14.genscan.txt | \sed 's/\.[0-9][0-9]$//' > NT_00512014.genscan.gff
Per FGENESH:
- $ gawk 'BEGIN{OFS="\t"} $4 ~ /CDSf|CDSi|CDSl/ {print "NT_005120.14", "fgenesh", $4, start=($5<$7 ? $5 : $7), end=($7<$5 ? $5 : $7), $8, $2, ".", $3, $1;}' NT_005120.14.fgenesh.txt | \sed 's/\.[0-9][0-9]$//' > NT_00512014.fgenesh.gff
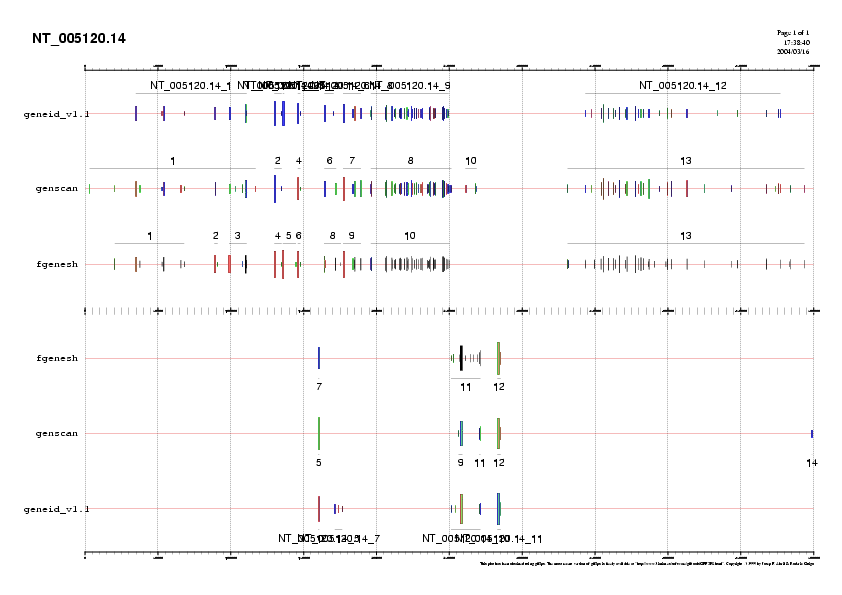
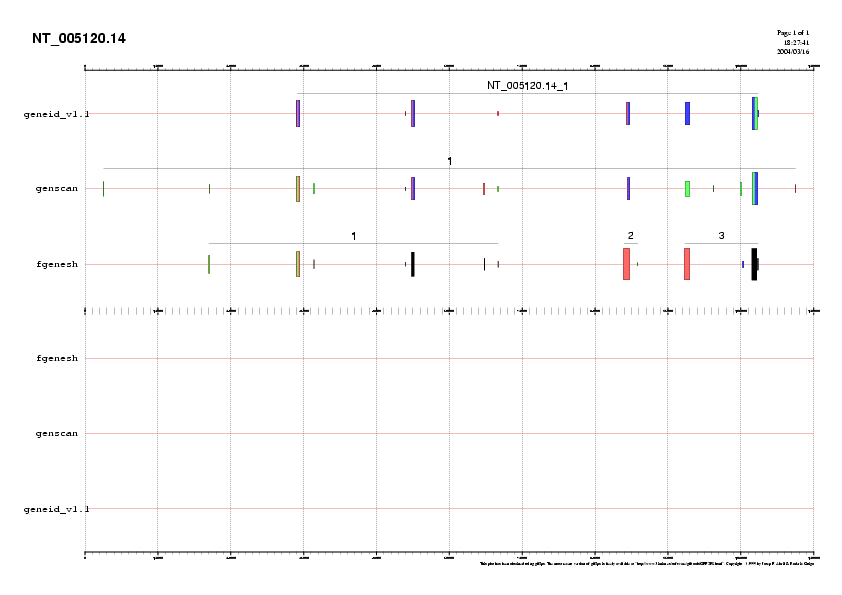
Un cop tenim tots els arxius en format .gff, ja els podem visualitzar conjuntament (Fig. 3a):
- $ gff2ps NT005120.14.geneid.gff NT005120.14.genscan.gff NT005120.14.fgenesh.gff > NT005120.14.genepredictions.ps
- $ convert -antialias -rotate 90 NT005120.14.genepredictions.ps NT005120.14.genepredictions.png
- $ kview NT005120.14.genepredictions.png
També generem un arxiu conjunt de les prediccions més els elements repetitius, de manera que podrem comprovar que allà on s'han predit exons no hi ha elements repetitius (Fig. 3b).
Ho fem seguint les següents comandes:
- $ gawk 'BEGIN{OFS="\t"} {print $5, $2="Repeat Masker",".", $6, $7, $1;}' repeat.seq.out > repeat.singleline.gff
- $ gff2ps NT_005120.14.geneid.gff NT_005120.14.genscan.gff NT_005120.14.fgenesh.gff repeat.singleline.gff > NT_005120.14.repeat+genepredictions.ps
- $ convert -antialias -rotate 90 NT_005120.14.repeat+genepredictions.ps NT_005120.14.repeats+genepredictions.png
- $ kview NT_005120.14.repeat+genepredictions.png
| a |
b |
 |
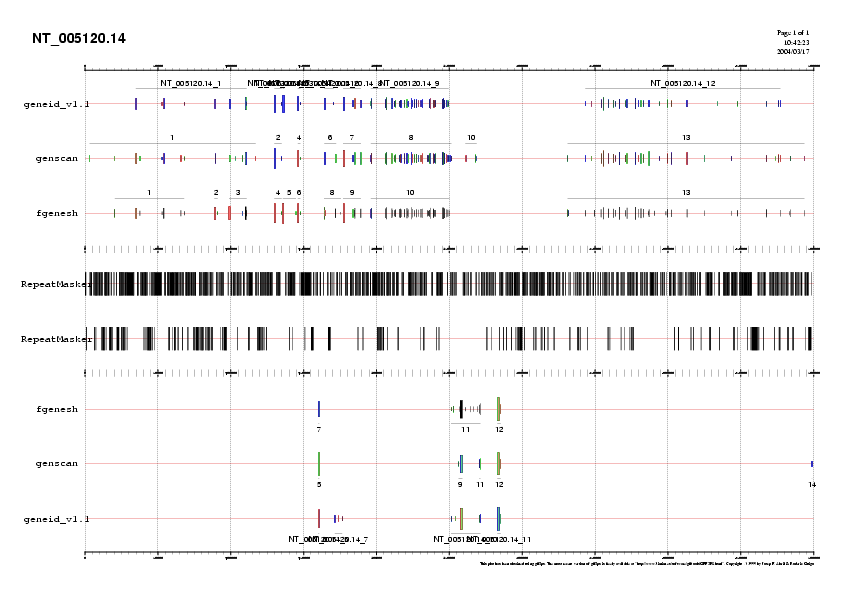 |
Fig.3 a Distrbució comparada dels possibles gens en la seqüència NT_005120.14 utilitzant diferents programes de predicció de gens.
b Mateixa predicció tenint en compte els elements repetitius.
Les prediccions de gens 'ab initio' donen resultats no del tot coincidents. La predicció amb GENEID dóna 92 exons agrupats en 12 gens; GENSCAN dóna 110 exons i 14 gens i FGENESH, 112 exons i 13 gens (Taules 2, 3 i 4).
Gens
predits |
Núm
exons |
Coordenades |
Forward(F)/
Reverse(R) |
| 1 |
8 |
34904-110908 |
F |
| 2 |
2 |
129814-134895 |
F |
| 3 |
1 |
135643-136707 |
F |
| 4 |
2 |
145949-147887 |
F |
| 5 |
2 |
160063-160738 |
R |
| 6 |
2 |
164164-170625 |
F |
| 7 |
3 |
171417-176520 |
R |
| 8 |
5 |
177110-189381 |
F |
| 9 |
35 |
196181-249474 |
F |
| 10 |
5 |
251233-271301 |
R |
| 11 |
2 |
282947-285266 |
R |
| 12 |
25 |
343296-477235 |
F |
|
Taula 2. Característiques dels gens predits per GENEID.
Podem observar el nombre de gens que aquest programa ens ha predit,
a l'igual que el nombre d'exons per a cada un, l'inici i final del
gen i si es troba en la cadena de forward o reverse.
|
Gens
predits |
Núm
exons |
Coordenades |
Forward (F)/
Reverse (R) |
| 1 |
14 |
3054-117025 |
F |
| 2 |
2 |
129814-134895 |
F |
| 3 |
1 |
135643-136707 |
F |
| 4 |
2 |
145949-147887 |
F |
| 5 |
2 |
160063-160738 |
R |
| 6 |
2 |
164164-172292 |
F |
| 7 |
5 |
177110-189381 |
F |
| 8 |
40 |
196181-251362 |
F |
| 9 |
2 |
256339-258929 |
R |
| 10 |
4 |
260944-268742 |
F |
| 11 |
2 |
270586-271301 |
R |
| 12 |
2 |
282947-285266 |
R |
| 13 |
31 |
331054-493689 |
F |
| 14 |
1 |
498805-499017 |
R |
|
Taula 3. Característiques dels gens predits per GENSCAN.
Es pot observar el nombre de gens predits i el nombre d'exons que
els formen, les coordenades d'inici i final del gen i si es troba en
en la cadena de forward o reverse.
|
Gens
predits |
Núm
exons |
Coordenades |
Forward (F)/
Reverse (R) |
| 1 |
7 |
20375-68085 |
F |
| 2 |
2 |
88757-91060 |
F |
| 3 |
4 |
98760-110908 |
F |
| 4 |
2 |
129814-134895 |
F |
| 5 |
2 |
135643-144791 |
F |
| 6 |
2 |
145949-147887 |
F |
| 7 |
2 |
160063-160738 |
R |
| 8 |
4 |
164036-175547 |
F |
| 9 |
5 |
177110-189381 |
F |
| 10 |
37 |
196181-250081 |
F |
| 11 |
10 |
251233-271301 |
R |
| 12 |
2 |
282947-285266 |
R |
| 13 |
33 |
331065-493689 |
F |
|
Taula 4. Característiques dels gens predits per FGENESH.
El programa ens ha predit el nombre de gens i exons que els formen,
l'inici i final de cadascun i si es troba en forward o reverse.
|
VALIDACIÓ DE LA PREDICCIÓ
Els programes de predicció de gens permeten determinar les zones que tenen major probabilitat de ser codificants, però s'hauran de validar els resultats obtinguts, quelcom que farem comparant amb una base de dades d'ESTs humans. Els ESTs (Expressed Sequence Tags) són fragments de cDNA obtinguts a partir de mRNA madur, que per tant representen regions codificants del genoma. Si aliniem el nostre fragment amb una base de dades de ESTs humans, podrem trobar quines regions de la nostre seseqüència s'hi assemblen, és a dir, quines regions són codificants i per tant validar els resultats obtinguts anteriorment.
Per millorar la qualitat d'aquesta validació dividirem la nostra seqüència en diversos blocs, que englobin possibles gens. Observant el gràfic (Fig. 4) distingim 4 regions riques en seqüències codificants, que determinen possibles gens. Amb la següent comanda dividirem l'arxiu:
- $ fastachunk repeat.seq.masked 0 125000 | fold -60 > subseqA.fa
on el 0 representa la posició des d'on volem tallar i el 125000 el nombre de nucleòtids que volem seleccionar. Obtenim així les següents subseqüències:
Taula 5 Subseqüències escollides.
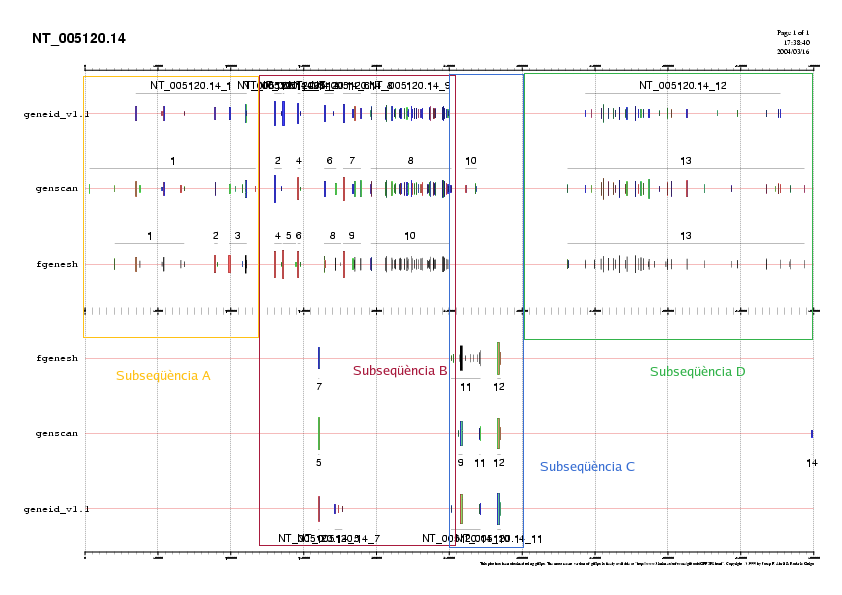
Fig. 4 Representació de les subseqüències
Un cop definits els diferents subarxius passem a buscar els ESTs presents mitjançant el BLASTN. Els BLASTN dóna uns arxius de sortida que anomenem subseq*.blastn.txt (utilitzarem el símbol * per A, B, C o D). Aquests han de ser manipulats per poder-se visualitzar, això ho farem utilitzant el programa gff2ps. Per poder-lo utilitzar hem de dur a terme una conversió del nostre arxiu subseq*.blastn.txt a subseq*.blastn.gff (com per exemple el subseqA.blastn.txt), quelcom que aconseguirem, utilitzant un script de perl i unes comandes:
- $ /disc8/bin/parseblast.pl -Gi subseq*.blastn.txt | gawk 'BEGIN{ OFS="\t" }{ $2=$9; $6=$7="."; print }' > subseq*.blastn.gff
- $ gff2ps subseq*.blastn.gff > subseq*.blastn.ps
- $ gawk -f getspliced.awk subseq*.blastn.gff > subseq*.blastn.spliced.gff
Podriem visualitzar els arxius subseq*.blastn.spliced.gff, però obtindriem uns gràfics que serien difícils d'interpretar per la gran quantitat de dades redundants, per això filtrarem i eliminarem els ESTs redundants:
- $ sort +3n -5 -o subseq*.blastn.spliced.gff subseq*.blastn.spliced.gff
- $ gawk '{exon[$4" "$5]=$9}END{for (i in exon) print exon[i];}' subseq*.blastn.spliced.gff | sort | uniq > subseq*.blastn.splicedord.gff
- $ gawk '{
if ($4" "$5 in sco) {
sco[$4" "$5]+=1;
} else {
sco[$4" "$5]=1;
};
exon[$4" "$5" "NR]=$0; printf "#%s", NR;
}
END{ printf "\n";
for (i in exon) {
split(exon[i],m," ");
print m[1],"blastn",m[3],m[4],m[5],sco[m[4]" "m[5]],m[7],m[8],m[9];
};
}' subseq*.blastn.spliced.gff | sort +8 -9 +3n -5 > subseq*.blastn.splicedresc.gff
- $ grep -f subseq*.blastn.splicedord.gff subseq*.blastn.splicedresc.gff > subseq*.blastn.splicedrescord.gff
Preparem 4 arxius amb les prediccions fetes per GENEID, GENSCAN i FGENESH per cada bloc i que anomenem NT_005120.14.genepredictions*.gff. Aquests arxius els podem passar a .png i visualitzar-los (llavors veuriem l'agrupació dels diferents exons formant gens) o unir-los a l'arxiu d'ESTs per poder comparar la predicció feta pels programes i els ESTs coneguts.
- $ gawk '$4>0 && $5<125000{OFS="\t"; $4=$4-0+1;$5=$5-0+1;print }' NT_005120.14.geneid.gff NT_005120.14.genscan.gffNT_005120.14.fgenesh.gff > NT_005120.14.genepredictionsA.gff
- $ gff2ps subseq*.blastn.splicedordrescord.gff NT_005120.14.genepredictions*.gff > subseq*.est+genepredictions.ps
- $ convert -antialias -rotate 90 subseq*.est+genepredictions.ps subseq*.est+genepredictions.png
- $ kview subseq*.blastn.png
Obtenim els resultats que es mostren en els esquemes de la Fig. 5, 6, 7, 8:
| Subseqüència A |
Subseqüència A amb ESTs |
 |
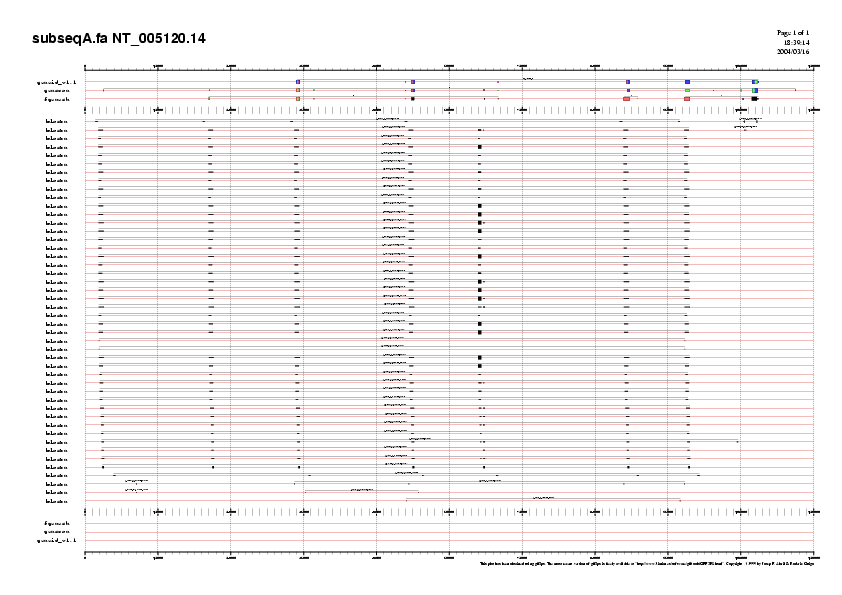 |
Fig.5. Subseqüències predites sense i amb ESTs.
GENEID prediu un gen de 8 exons; GENSCAN prediu un gen de 14 exons; FGENESH prediu 3 gens de 7, 2 i 4 exons, tots ells en forward. La llargada dels gens predits és diferent. Per GENEID obtenim un gen de 76004 pb, per GENSCAN dóna un gen més llarg, de 113971 pb, amb 2 exons addicionals en 5' i un en 3'. FGENESH prediu 3 gens (47710, 2303, 12148) el primer dels quals comença en el segon exó predit per GENSCAN. Els ESTs validen tots els exons predits per GENSCAN, a excepció de l'últim (que tampoc no apareix en les prediccions per GENEID i FGENESH). Utilitzarem el gen 1 de GENSCAN per validar la predicció comparant-la amb amb una base de dades de proteïnes.
| Subseqüència B |
Subseqüència B amb ESTs |
 |
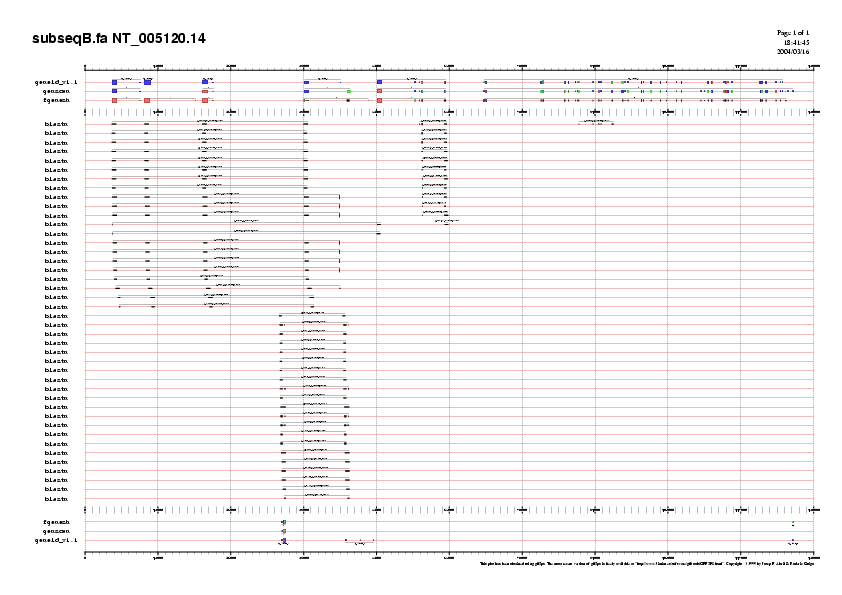 |
Fig.6. Subseqüències predites sense i amb ESTs.
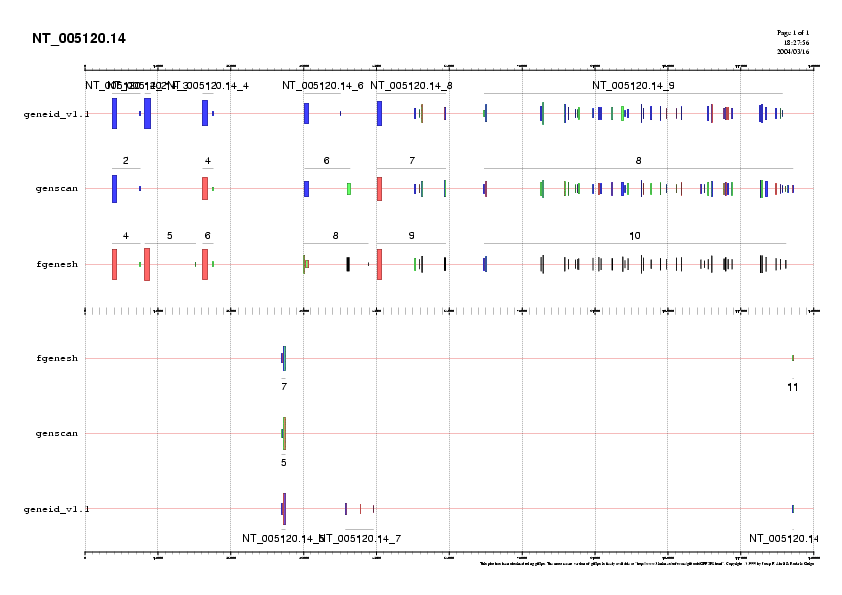
GENEID prediu 6 gens de 2, 1, 2, 2, 5 i 35 exons en forward, i 2 gens de 2 i 3 exons; en reverse. GENSCAN prediu 5 gens de 2, 2, 2, 5 i 40 exons en forward, i un gen de 2 exons en reverse. FGENESH prediu 6 gens de 2, 2, 2, 4, 5 i 37 exons en forward, i un gen de 2 exons en reverse (els gens 14 de GENEID i 11 de FGENESH corresponen a la subseqüència C). És destacable la falta d'un exó en la predicció per GENSCAN (corresponent als gens 2 de GENEID i 5 de FGENESH).
El primer que s'observa a la Fig.6 b, és el fet de que les prediccions 9 de GENEID, 8 de GENSCAN i 10 de FGENESH (bastant coincidents entre elles), curiosament, no estan suportades pels ESTs. Únicament apareix un EST recolzant 5 exons interns del gen. Com que els tres programes han predit un gen llarg amb molts dels exons coincidents, creiem que és probable la presència d'un gen transcrit en aquesta regió, malgrat l'absència de ESTs. Per verificar si es tracta de la mateixa proteïna, compararem les tres prediccions entre elles mitjançant el programa ClustalW. Aquesta regió pasarema anomenar-la subseqüència B2.
Per als gens 8 GENEID, 7 GENSCAN i 9 FGENESH, tenim ESTs recolzant els tres últims exons.
Per a la regió dels primers 25000 pb els ESTs suporten els exons més grans de les prediccions, però, a diferència del que fan els programes de predicció de gens, els ESTs ajunten aquests exons en un sol gen. Veiem que els ESTs també ajunten els exons localitzats en els següents gens predits (6 de GENEID i GENSCAN i 8 de FGENESH). Creiem que això és incompatible amb la presència d'un gen en reverse que prediuen els tres programes i els exons dels quals es troben recolzats per ESTs (gen 6 de GENEID, 5 de GENSCAN i 7 de FGENESH). Si en un inici pensavem que teniem gens diferents (degut a la presència del gen en reverse), veient que els ESTs ens els uneixen no podem estar segures, per tant haurem de fer un estudi detallat de cada fragment.
Considerem per tant que aquesta subseqüència podria contenir 3 gens diferents en forward o 4 en forward i 1 en reverse. Donat que les prediccions són confuses tractarem de validar una de les dues hipòtesis estudiant les següents proteïnes (indicada la predicció de la que farem el BLASTP):
- fragment de 0-25000:
- Gen B1e, gen 2 de GENSCAN
- Gen B1d, gen 4 de GENSCAN
- Gen en reverse: Gen B1a, gen 7 de FGENESH
- Gen B1c, gen 6 de GENSCAN
- Gen B1b, gen 7 de GENSCAN
- Gen B2, gens 9 de GENEID, 8 de GENSCAN i 10 de FGENESH
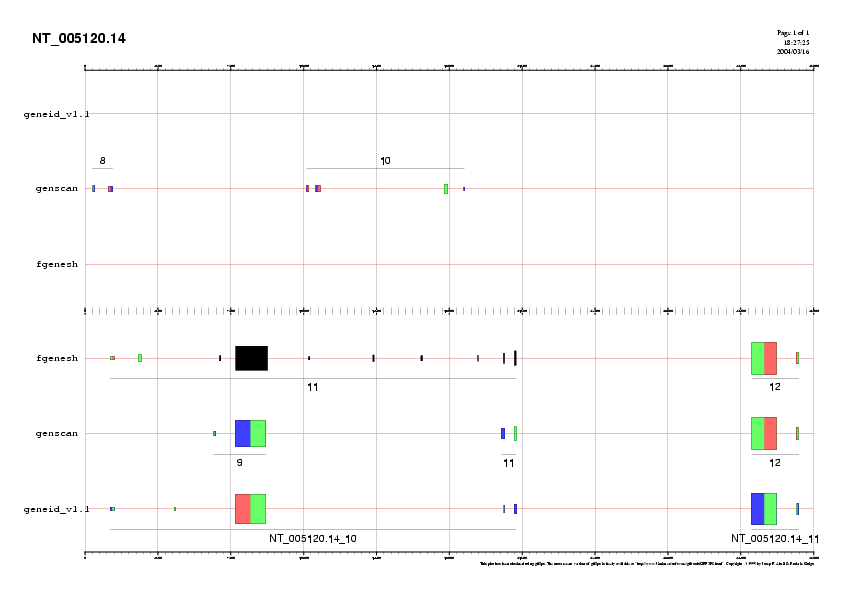
| Subseqüència C |
Subseqüència C amb ESTs |
 |
 |
Fig.7. Subseqüències predites sense i amb ESTs.
GENEID prediu 2 gens de 5 i 2 exons en reverse i un gen de 4 exons en forward. GENSCAN prediu 3 gens de 2, 2 i 2 exons en reverse. FGENESH prediu 2 gens de 10 i 2 exons en reverse. En la regió dels primers 30000 pb FGENESH i GENEID prediuen un sol gen en reverse mentre que GENSCAN en prediu 2 en reverse separats per un gen en forward. Els ESTs suporten majoritàriament la predicció feta per FGENESH i afegeixen un exó addicional entre els exons 5 i 6 del gen 11. Els ESTs també suporten el segon exó del gen 10 de GENEID. Les prediccions pels gens 12 de FGENESH i GENSCAN i 11 de GENEID (de 2 exons) són molt consistents entre elles i es validen amb els ESTs.
Per la caracterització de les proteïnes, agafarem per una banda la proteïna predita del gen 11 de FGENESH i per l'altra el gen 12 també de FGENESH.
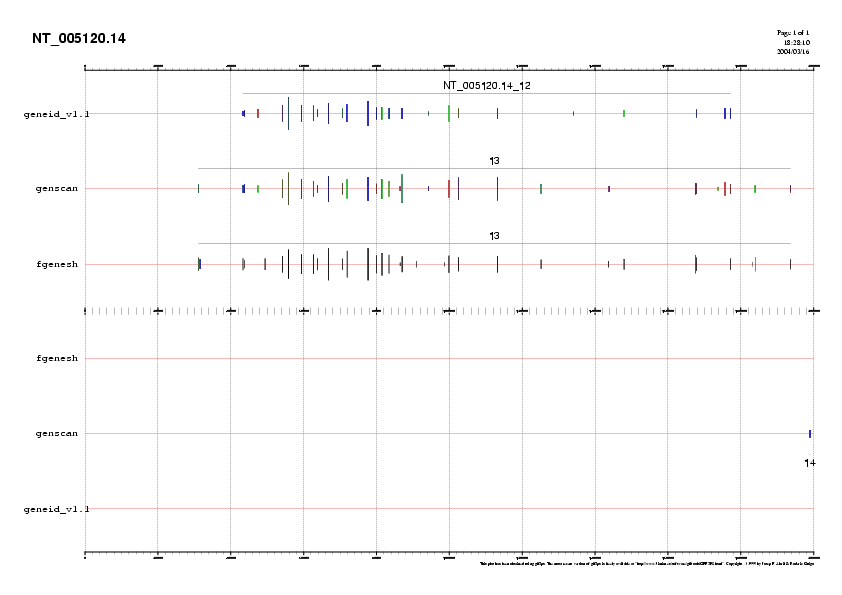
| Subseqüència D |
Subseqüència D amb ESTs |
 |
 |
Fig.8. Subseqüències predites sense i amb ESTs.
Els tres programes prediuen en aquesta regió un sol gen llarg en forward. GENEID prediu 1 gen de 133939 pb i 25 exons. GENSCAN prediu un gen de 162635 pb i 31 exons. FGENESH prediu un gen de 162624 pb i 33 exons. GENEID no prediu exons en 5' i 3' que sí prediuen els altres dos programes.
Els ESTs suporten els 5 ültims exons de la prdició per FGENESH (últimes 50000 pb). A més els ESTs suporten l'exó addicional que predeien GENEID i GENSCAN i també introdueixen un exó de més (entre els dos últims exons de FGENESH). Respecte la resta de subseqüència, els ESTs suporten només en part els exons. Els 3 primers exons predits per GENSCAN i els 4 primers de FGENESH no es troben suportats per cap EST. Tampoc no tenim cap EST pels exons presents en 140000-150000 pb de la subseqüè. A més trobem ESTs amb exons addicionals en la regió 120000-140000.
Donat el fet que la predicció és poc consistent en la regió de 120000-150000 pb és possible que la subseqüència contingui, com a mínim 2 gens, separats per aquests 30000 pb. Tractarem de confirmar aquesta hipòtesi comparant una de les prediccions amb la base de dades de proteïnes. Utilitzarem la predicció de GENSCAN perquè, de les tres, és la que més s'ajusta a la predicció per ESTs.
ANÀLISI DE LES PROTEÏNES PREDITES
Per tal de validar la presència de regions codificants en la seqüència, comparem de les proteïnes predites pels programes GENEID, GENSCAN i FGENESH, les triades amb la base de dades de proteïnes humanes de SwissProt mitjançant el programa BLASTP.
Subseqüència A
S'ha utilitzat la proteïna corresponent al gen 1 del GENSCAN i s'obtenen els següents resultats:
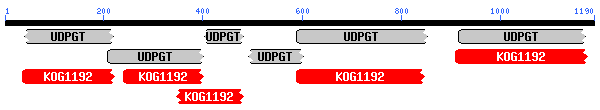
Fig.9 Dominis conservats
BLASTP A
En l'esquema de dominis conservats obtenim mutiples fragments d'un domini UDP-glucoronosyltransferase i UDP-glucosyltransferase. Els resultats obtinguts amb el BLASTP mostren diverses entrades, les primeres 18 pertanyents a la familia de la UDP-glucuronosyltransferases (UGT1), i amb molt baix score. Donat que es tracta de proteïnes de la mateixa famïlia, amb gran homologia entre elles, no podem saber de quina de totes es tracta.
Subseqüència B
Proteïna B1a (en reverse)
S'ha utilitzat la proteïna codificada pel gen 7 de FGENESH, aconseguim els següents resultats:

Fig.10 Dominis conservats
BLASTP B1a
Amb l'estudi de dominis obtenim dominis conservats de DNA-J chaperone i els resultats obtinguts amb el BLASTP alineen el gen amb una proteïna DnaJ homolog subfamily B member 6, amb un score de 8e-56, validant els resultats anteriors.
Proteïna B1b
S'ha utilitzat la proteïna codificada pel gen 7 de GENSCAN, aconseguint els següents resultats:
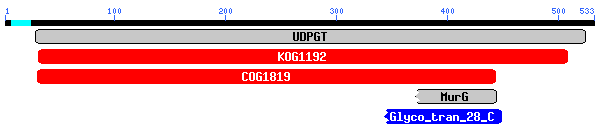
Fig.11 Dominis conservats
BLASTP B1b
S'obté un domini sencer conservat corresponent a UDP-glucoronosyltransferase i UDP-glucosyltransferase amb un e-value de 0.0. També s'obtenen fragments C-terminals de dominis MurG UDP-N-acetylglucosamine i Glycosyltransferase family 28, amb e-values majors. Els resultats de BLASTP tornen a donar múltiples alineaments amb UDP-glucoronosyltransferases (UGT1), amb uns e-values de 0.0.
Proteïna B1c
S'ha utilitzat la proteïna codificada pel gen 6 de GENSCAN, i s'obté el següent:
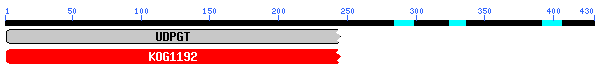
Fig.12 Dominis conservats
BLASTP B1c
Tornem a obtenir dominis conservats UDP-glucoronosyltransferase i UDP-glucosyltransferase (fragment N-terminal). El BLASTP mostra de nou múltiples alineaments amb proteïnes de la família de les UDP-glucoronosyltransferases (UGT1).
Proteïna B1d
Utilitzem la proteïna codificada pel gen 4 de GENSCAN, i s'obté el següent:

Fig.13 Dominis conservats
BLASTP B1d
De nou l'esquema de dominis conservats mostra un fragment N-terminal d'un domini UDP-glucoronosyltransferase i UDP-glucosyltransferase, amb un e-value de 2e-89. En el cas del BLAST tornem a trobar múltiples alineaments significatius amb membres de la família de les UGT1.
Proteïna B1e
Utilitzem la proteïna codificada pel gen 2 de GENSCAN, obtenint els següents resultats:

Fig.14 Dominis conservats
BLASTP B1e
Novament apareix un domini conservat UDP-glucoronosyltransferase i UDP-glucosyltransferase, amb un e-value de 3e-83. Els resutats del BLASTP tornen a donar alineaments amb proteïnes de la famïlia UGT1, amb e-values molt baixos.
Els resultats dels BLASTPs de les proteïnes predites A (3054-117025), B1e (129814-134895), B1d (145949-147887), B1c (164164-172292), B1b (177110-189381), localitzades totes elles en les primeres 190000 pb, indiquen que són proteïnes de la família UDP-glucoronosyltransferase 1 (UGT1) i mostren un alt grau d'homòlogia entre elles. També observem que totes aquestes proteïnes s'alineen amb dominis N-terminals a diferents alçades, i únicament la proteïna predita B1b s'alinea amb el domini C-terminal. Amb aquestes dades podriem pensar que aquest fragment, que els programes de predicció de gens han dividit en diversos gens, és un únic gen que pateix splicing alternatiu. Aquesta hipòtesi es veuria suportada pels ESTs que ajuntaven tot aquest fragment en un únic transcrit. La presència, però, de la proteïna B1a en reverse i localitzada entre la B1c i B1d, ens fa dubtar d'aquest hipòtesi.
Deguda l'alta complexitat d'aquest fragment buscarem informació sobre la UGT1. A partir de l'article Multiple Variable First Exons: A Mechanism for Cell- and Tissue-Specific Gene Regulation. Zhang et al. sabem que aquest fragment correspon a un cluster de gens de la UGT1, proteïnes que entre elles es diferencien únicament pel domini N-terminal. Aquest cluster conté una regió variable d'exons amb alta homologia entre ells disposats en tandem, una regió constant de quatre exons, i exons que pertanyen a pseudogens (UGT1 A11p, A12p, A13p, A2p) i a altres proteïnes no homòlogues, com la Hsp40-DnaJ like. Aquest cluster de gens no és un sol gen que pateix splicing alternatiu com semblaven suportar els ESTs, sinó un conjunt de gens amb una regulació poc usual en què cada exó de la regió variable es troba sota control d'un promotor propi.
Amb aquestes dades veiem que el que els programes de predicció prediuen com el gen 7 de GENSCAN probablement correspon a la regió constant del cluster, de 4 exons (domini C-terminal) més un altre exó en N-terminal, pertanyent a UGT1A1; mentre que el gen 7 predit per FGENESH, que els nostre resultats alineava amb la DnaJ, segurament correspon a Hsp40. En conjunt veiem que la regió de les primeres 190000 pb correspon al cluster de la UGT1(Fig. 15).
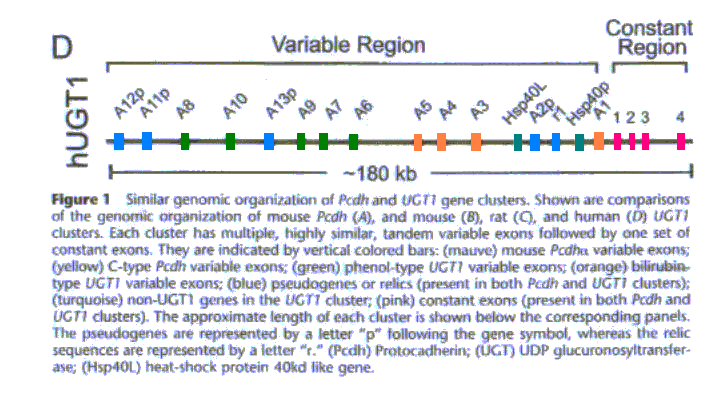
Fig.15 UGT1 cluster. En verd i taronja veiem els diferents exons de la part variable d'aquest cluster. En blau estan representats els pseudogens; en turquesa exons de gens que no pertanyen al cluster UGT1; i en rosa els exons de la part constant del cluster.
Proteïna B2
Donat que els resultats dels 3 programes de predicció de gens eren bastant coincidents entre ells i, en canvi, els ESTs no els recolzen, fem un alineament múltiple de les 3 proteïnes predites amb el programa ClustalW per tal de confirmar que es tracta de la mateixa proteïna. És possible que no existeixin ESTs seqüenciats per aquesta regió i que per tant no estiguin a la base de dades.
Fem ClustalW amb:
- La proteïna 9 de GENEID (1556 aa)
- La proteïna 8 de GENSCAN (1731 aa)
- La proteïna 10 de FGENESH (1588 aa)
L'alineament obtingut teacute; una puntuació molt alta. La porció N-terminal, fins l'aminoàcid 531, obté un alineament total de les tres proteïnes. Fins l'aminoàcid 1592 de la predicció: de GENSCAN, 1548 de FGENESH i 1531 de GENEID obtenim alguns gaps en l'alineament, però els aa alineats són idèntics entre ells (podem veure els resultats aquí ). Podem concloure, per tant, que les prediccions són molt similars entre elles i que podem utilitzar qualsevol de les tres proteïnes predites per fer el BLASTP. Utilitzarem la predicció de GENSCAN, la més llarga
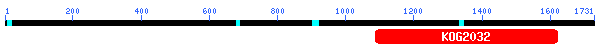
Fig.16 Dominis conservats
BLASTP B2 SwissProt
Com podem veure als resultats, el BLASTP de protB2 contra la base de dades SwissProt no dóna cap hit significatiu. En canvi, sí que trobem un domini conservat (KOG2032) de funció desconeguda en la regio 1100-1600 aa de la proteina Això és consistent amb el fet que no hi hagin aparegut ESTs alineats en aquesta regió. És possible que la proteïna no estigui caracteritzada i que per tant no existeixi a la base de dades SwissProt. Per confirmar l'existència d'un transcrit farem un nou BLASTP però aquest cop contra tota la base de dades (opció Database = nr).
BLAST B2 nr
Amb el nou alineament obtenim dos hits significatius (e-value = 0.0) per humà, corresponents a:
- hypothetical protein XP_291007
- similar to hypothetical protein FLJ40243 (cromosoma 2q37)
La hipòtesi, doncs, d'una proteïna no caracteritzada és consistent amb les dades donat que són proteïnes hipotètiques. Tot i que ambdues proteïnes donen un e-value de 0.0, la que més probablement correspon a la nostra proteïna predita és similar to hypothetical protein FLJ40243, donada la seva localització.
Subseqüència C
Proteïna C1
Utilitzem la proteïna codificada pel gen 2 de GENSCAN, obtenint els següents resultats:
BLASTP C1
L'alineament de la proteïna contra la base de dades SwissProt no dóna cap hit significatiu. Degut a l'alt suport d'ESTs per als exons, decidim alinear la nostra proteïna contra tota la base de dades. Mostrem els resultats en el següent link BLASTP. Amb el nou alineament obtenim dues proteïnes amb alta homologia amb la nostre seqüència (e-value 0.0), aquestes són:
- unamed protein product (BAC11221)
- hypothetical protein DKFZp762E1312 (cromosoma 2q37)
Tenint en compte que la unamed protein és un BAC, i que la hypotetical protein es troba al cormosoma 2q37, el més possible és que sigui aquesta la que es trobi codificada per aquest gen.
Proteïna C2
Per l'estudi d'aquest fragment vam utilitzar el gen 12 de FGENESH, obtenint-se:
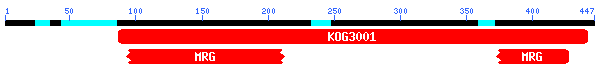
Fig.17 Dominis conservats
BLASTP C2
Els resultats mostren un domini Dosage compensation regulatory complex/histone acetyltransferase complex, amb un e-value de 1e-76, i dos fragments d'un domini MRG (que comprén 3 proteïnes, mortality factor 4, male-specific lethal 3 i ESA1-associated factor 3), amb e-values majors. Els resultats del BLAST mostren un alineament bastant òptim amb la Male-specific lethal 3-like 1 (e-value de e-160). Tenint en compte les dues dades podem deduir que la proteïna predita es tracta de la Male-specific lethal 3-like 1.
Subseqüència D
Per acabar amb l'estudi vam utilitzar el gen 13 de GENSCAN, i trobem:
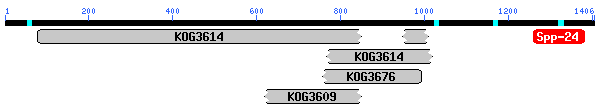
Fig.18 Dominis conservats
BLASTP D
Els resultats mostren que la proteïna conté a la seva regió N-terminal (primers 1000 aa) fragments d'un domini Ca2+/Mg2+-permeable cation channels, amb un e-value de 0.0. I en la porció C-terminal un domini conservat en Secreted phosphoprotein 24 amb un e-value de 4e-31. El BLASTP mostra alta homologia amb una Long transient receptor potencial channel 2/7 (e-value e-165) en els primers 1000 aa, i amb un e-value de 6e-40 en la regió C-terminal trobem homologia amb la Secreted phosphoprotein 24, coincidint amb els resultats anteriors, el que ens fa pensar que efectivament la regió contenia dues proteïnes en lloc d'una, en l'extrem C-terminal una Secreted phosphoprotein 24 i en el N-terminal una Long transient receptor potencial channel 2/7.
CONCLUSIONS
L'objectiu del projecte és caracteritzar la seqüència ENr131 del projecte ENCODE, utilitzant diverses eines computacionals.
Tenint en compte els diferents resultats obtinguts per cada tipus d'estudi, podem resumir que en aquest fragment de DNA trobem 1 cluster de gens i 6 proteïnes (Fig 18). Així doncs tenim:
- UGT1 cluster (Subseqüències A, B1e, B1d, B1c, B1b)
- Hsp40-DnaJ like (en reverse) (Subseqüència B1a)
- Hypotetical protein FLJ402443 (Subseqüència B2)
- Hypotetical protein DKFZp762E1312 (en reverse) (Subseqüència C1)
- Male-specific lethal 3-like 1 (en reverse) (Subseqüència C2)
- Long transient receptor potencial channel 2/7 (LTrpC2/7) (Subseqüència D)
- Secreted phosphoprotein 24 (Spp24) (Subseqüència D)
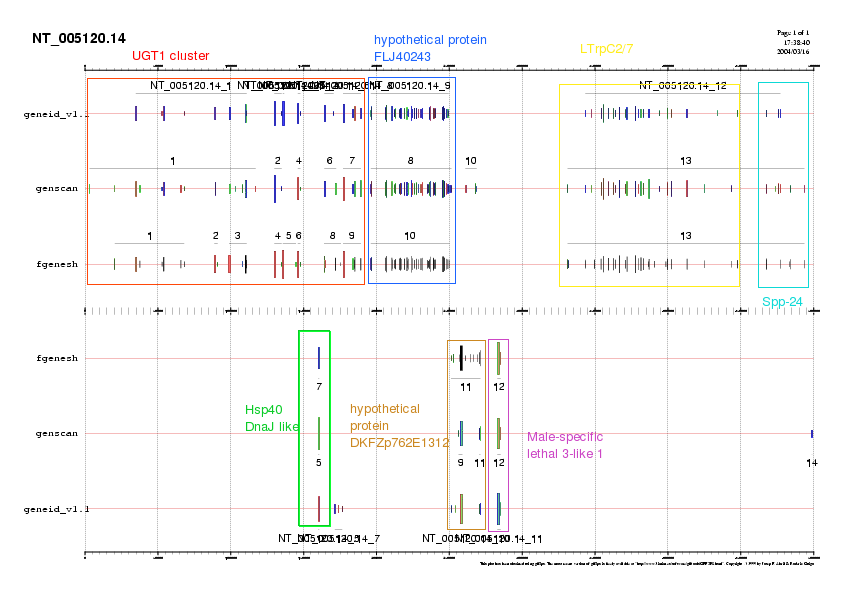
Fig. 19 Esquema de la localització dels gens.
Veient els resultats podem dir que els programes de predicció de gens que hem utilitzat no són del tot eficaços, ja que han fet prediccions diferents, amb exons i gens diferents i en desacord amb els ESTs. Val a dir, però, que la regió sembla tenir una alta densitat gènica i una estructura complexa (pel que fa al cluster UGT1) cosa que dificulta una correcta predicció dels gens.
AGRAÏMENTS
Volem agrair molt especialment el suport de les companyes d'anàlisi
de seqüències, durant aquesta intensiva etapa de la nostra vida.
També agraïr l'ajuda prestada pels tutors.
I no oblidem la bona fe de les nostres mares/pares per preparar-nos el
tupper tots aquest dies agònics. |
|
|
 |
GRÀCIES!!!!
Si necessiteu contactar amb nosaltres, si us plau, no deixeu d'enviar un mail a una de les següents direccions:
Lidia Tubert
Míriam Onrubia
BIBLIOGRAFIA
