3. Project
The aim of this project is to find out novel U12 introns in the human genome.To get this, we have had to apply some PERL programming skills by parsing GFF files, extracting sequences from the genome and organize our results.
GeneID has been modified to be able to detect U12splice sites and assemble exons with U12 donor or acceptor sites into valid gene models (Geneid_v1.2_u12). Although it is very sensitive, its specificity is still low. Then, Gene ID intron predictions require a post-processing process.
|
WORK PLANNING
We started from two U12-intron-flanking exons predictions (gff files), one obtained from the Geneid_v1.2_u12 (GeneIDU12_hg17.gff
) and the other one from Syntenic Gene Prediction, SGP (SGPU12_hg17.gff). We had to get the introns derived from those files. To do this, we wrote a script (exons2introns.pl). This program may be useful to look for all the predicted U12 introns flanked by two U12-intron-flanking exons (atac or gtag variants). After this step, we applied our script twice, one for the GeneID predictions and another for the SGP predictions. So the output consists of two gff files with predicted U12 introns: intronsIDU12.gff and intronsSGPU12.gff. Once we had this information, and using some UNIX commands, we mixed both gff files removing the repeated predicted U12 introns. The command we used to do this was:
cat intronsIDU12.gff intonsSGPU12.gff |sort -u -k1,1 -k4,5n As a result, we get a new gff file with 2064 predicted U12 introns (intronsU12pred.gff). As our supervisor had already predicted and corroborated some of the U12 introns that we had in the previous file, we combined our predictions with his file which contains 648 real U12 introns (GeneID_SGP2_LD_ESTconf_know.gff). We applied gffintersect.pl: a script that finds intersection of two GFF files; prints lines from file2 that intersect (or do not intersect) with file1. Applying this script to intronsU12pred.gff and GeneID_SGP2_LD_ESTconf_know.gff we get three new gff files:
|
perl gffintersect.pl -minfrac1 -minfrac2 -quiet intronsU12pred.gff GeneID_SGP2_LD_ESTconf_know.gff --> gffintersectC.gff
perl gffintersect.pl -not -minfrac1 -minfrac2 -quiet intronsU12pred.gff GeneID_SGP2_LD_ESTconf_know.gff --> gffintersectK.gff
perl gffintersect.pl -not -minfrac1 -minfrac2 -quiet GeneID_SGP2_LD_ESTconf_know.gff intronsU12pred.gff --> gffintersectP.gff
|
it belongs to common lines between the files. So they are 86 real U12 introns found by our supervisor. We will use them as a positive control to support our results.
gffintersectK.gff: these are 562 known U12 introns that nor GeneID nor SGP had been able to predict.
gffintersectP.gff: these are 1978 U12 intron predictions that we have to test in order to determine which of them are real.
To measure U12intron-prediction accuracy, we used a search algorithm (BLAST) to find matching or similar sequences in dbEST (a division of GeneBank that contains sequence data and other information on 'single-pass' cDNA sequences, or Expressed Sequence Tags, from a number of organisms) and 'nr' database (the largest nucleotide database available through NCBI BLAST).
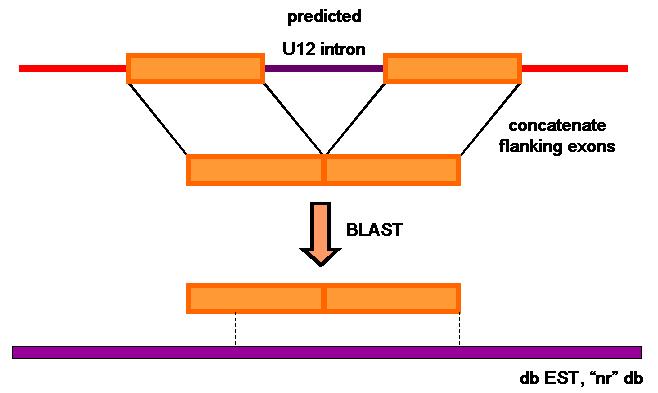
Our hypothesis consists on if we can find in those databases a significant match with the predicted U12-intron-flanking exons, it is because the predicted U12 intron really exists. So, before doing the BLAST, we should concatenate the flanking exons of every predicted U12 intron and obtain its nucleotide sequence. Instead of using all the sequence of the flanking exons, we used only the 50 pb nearer the precicted U12 intron. During the results analysis, BLAST output will be classified in different categories:
- Alignment across junction: both splice sites (exon flanks) coincide with EST and protein databases, including the junction between them. Complete support for intron prediction.
- Two exons: both splice sites (exon flanks) coincide with EST and protein data bases, but not the junction between them.
- One exon flank (right or left): at least one splice site (one exon flank) coincides with EST and protein databases. Partially supported.
- No-hits: no hits to any expressed sequence or protein. Completely unsupported.
In order to get the sequence of the flanking exons we wrote a second script (introns2sequence.pl
). By means of this, we can concatenate the sequence of the flanking exons that we need (50 pb of every exon) to do the BLAST. The nucleotide sequence is obtained from a human genome database provided by our supervisor. After applying the script we obtained two files that we used to do the BLAST analysis: exonpairsP50.fa and exonpairsC50.fa.
We only did the BLAST with our predicted U12 introns ('P' file) and with the common U12 introns ('C' file) that we will use as a positive control. We did not use 'K' file because we already know that they are real U12 introns and they are not predicted with GeneID or SGP.
BLAST (Basic Local Alignment Search Tool) is an algorithm for comparing biological sequences, such as the amino-acid sequences of different proteins or the DNA sequences. It emphasizes speed over sensitivity. It is important to remember that BLAST is a local alignment tool. We used two different kinds of BLAST:
- BLASTN: compares our nucleotide query sequence (exonpairs files) against an EST database.
- BLASTX: compares our nucleotide query sequence (exonpairs files) translated in all reading frames against a protein database ('nr').
Once we had done the BLAST, we obtained four new files which contain the results of the alignments against the 'nr' database and dbEST for the 'P' and 'C' files:
Using another script (woundedknee.pl
) we classified the predicted U12 introns into five different categories (possibilities of BLAST matching) : alignment across junction, two exons, one exon flank right, one exon flank left and no-hits. We compiled our results in a table. Finally, we searched in the UCSC genome browser some of the new U12 introns that we have found.