
 |  |  |  |  |  |  |
| INTRODUCCIÓN | OBJETIVOS | MATERIALES | RESULTADOS | DISCUSIÓN | AGRADECIMIENTOS | REFERENCIAS |
La doble función de codificación del codón UGA supone un importante reto en la anotación de genomas. A pesar de que UGA normalmente señala la terminación de la síntesis protéica, UGA también puede codificar para selenocisteína (Sec), la cual es incorporada en un pequeño pero importante grupo de proteínas, conocidas como selenoproteínas. Los programas standard de análisis de genes no pueden predecir si UGA codifica para Sec o para Stop4.
Cuando se secuencia un nuevo genoma los codones UGA se anotan automáticamente como un codón stop, lo cual supone una barrera para la búsqueda de selenoproteínas. Para anotar estas proteínas en un nuevo genoma hay que buscar regiones de este genoma correspondientes a elementos característicos de las selenopreteínas que se mantienen conservados en aquellos organismos que codifican para ellas. Para comprobar esta conservación es necesario realizar un estudio de similitud entre las secuencias de selenopreoteínas humanas, por ejemplo, y el genoma de la especie de interés, en nuestro caso, Pan troglodytes. Algunos de estos elementos que caracterizan a las secuencias codificantes de selenoproteínas son:
Nuestro proyecto consiste en la busqueda de secuencias codificantes para selenoproteínas en el genoma de Pan troglodytes a partir de las 25 selenoproteínas ya anotadas en el genoma de Homo sapiens.
Cuando buscamos homologías lo que realmente estamos buscando son secuencias con una función en común pero que no tienen porque estar realcionadas desde el punto de vista evolutivo. En general, lo que queremos identificar son los dominios funcionales de una secuencia desconocida basándonos en la similitud de esa secuencia a otras secuencias conocidas y así ser capaces de inferir su función. BLAST (Basic Local Alignment Search Tool) es un algoritmo que permite comparar tanto secuencias aminoacídicas como secuencias de DNA. Es uno de los programas bioinformáticos más ampliamente utilizados probablemente porque resuelve los problemas planteados de una forma en la que prevalece la rapidez sobre la sensibilidad. El hecho de conceder más importancia a la velocidad de resolución sobre la sensibilidad lo hace idóneo para extraer información de interés de las enormes bases de datos de genomas que ya están seceunciadas y disponibles.
En el caso del tBLASTn lo que comparamos es una secuencia aminoacídica (query, que en nuestro caso corresponde a la secuencia proteica de una de las 25 selenoproteínas humanas), respecto a una secuencia nucleotídica o una base de datos nucleotídica traducida simultáneamente a las 6 posibles pautas de lectura (subject, que en nuestro caso corresponde a la base de datos del genoma de Pan troglodytes). BLAST usa un algoritmo heurístico que busca alineamientos locales, por lo que permite establecer relaciones entre secuencias que únicamente comparten regiones aisladas de similaridad. Los parámetros de configuración del tBLASTn de Ensembl para llevar a cabo la predicción de las regiones de similaritud han sido:
Para visualizar el resultado de nuestro tBLASTn haz click aquí.
Con el tBLASTn hemos restringido las regiones de los potenciales genes que codifican para selenoproteínas en el genoma de Pan troglodytes. Diferentes fragmentos de las selenoproteínas han alineado con diferentes regiones del genoma de Pan troglodytes, sin embargo para nuestro objetivo sólo son de interés aquellos alineamientos en los que vemos alineadas Sec con Sec o Sec con Cys. El tBLASTn muestra la Sec codificada con el símbolo * , el mismo símbolo que se utiliza para codificar los codones Stop. Por lo tanto los alineamientos de interés mostrarán el siguiente patrón:
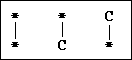
Para seleccionar únicamente aquellos alineamientos que contenían los emparejamientos arriba mencionados escribimos un programa en lenguaje perl, programa.pl
Con este programa extraímos las siguientes secuencias de interés.
Exonerate es un programa que alinea secuencias prediciendo cual podría ser la estructura exónica de la secuencia problema. Para llevar a cabo esta predicción Exonerate se basa tanto en heurísticos como en programación dinámica.
En nuestro caso hemos utilizado una versión (exonerate 0.8.2. model protein2genome) que alinea una secuencia proteica de Homo sapiens contra una secuencia nucleotídica del genoma de Pan troglodytes. Este modelo permite la presencia de intrones en el alineamiento pero también cambios en la pauta de lectura y cambios de fase en los exones cuando un codón es interrumpido por un intrón.
El programa lo hemos ejecutado desde la línea de comandos mediante las siguientes instrucciones:
$export PATH=$PATH:/disc8/bin/
exonerate --showtargetgff --model protein2genome query.fasta target.fasta
Donde:
-Input:
-Output: showtargetgff. Para que el exonerate nos devuelva el output en formato GFF.
-Model: protein2genome.
Para extraer únicamente la tabla gff del output del exonerate desarrollamos un programa en lenguaje perl programagff
Sólo para un caso en concreto, por sugerencia de nuestro tutor utilizamos GeneWise para predecir la secuencia exónica de la selenoproteína de Pan troglodytes. GeneWise al igual que hacíamos con Exonerate compara la secuencia proteica de Homo sapiens con la secuencia de DNA de Pan troglodytes, permitiendo la presencia de intrones y el cambio de pauta de lectura.
SECISearch 2.19, nos he permitido encontrar estructuras Secis en el extremo 3' de la selenoproteína predicha para Pan troglodytes. Lo que hicimos fue extrear una región de 1000 nucleotídos del extremo 3' de la secuencia genómica de la selenoproteina de Pan troglodytes, mediante Ensembl o UCSC, dependiendo de cual había sido la fuente de extracción de la misma.

Para porder hacer un alineamiento global entre la selenoproteína de Homo sapiens y la predicha para Pan troglodytes primero había que traducir a proteína los exones predichos por el Exonerate. Para la traducción utilizamos la herramienta de traducción que ofrece Expasy servidor.
Clustal W es un programa que hace alineamientos múltiples de secuencias biológicamente significativos, tanto de DNA como de proteína. Calcula la mejor coincidencia para las secuencias introducidas y pone una secuencia debajo de la otra de forma que muestra visualmente las coincidencias, similaridades y diferencias.
Gracias al alineamiento múltiple entre las selenoproteína de Homo sapiens y la selenoproteína predicha para Pan troglodytes hemos podido ver la conservición entre ambas.
![]()
![]()
