
SELENOPROTEÍNAS DE Meleagris gallopavo
MATERIALES Y MÉTODOS: MÓDULO IV
MAPA CONCEPTUAL
|
|||
|

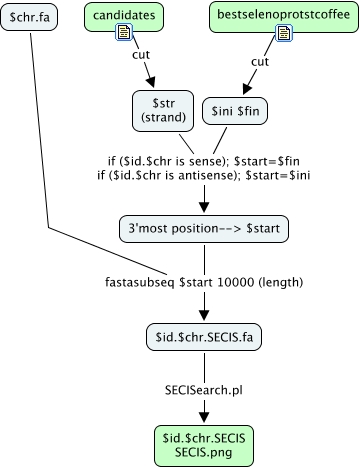
En este módulo hacemos una búsqueda de elementos secis en una región de 10kb a partir del extremo 3' de las mejores predicciones.
BASH script
#
###############################################################################################
################################## - MÓDULO 4 - ######################################
###############################################################################################
#################################### SECISearch ######################################
###############################################################################################
#!/bin/bash
#$ -o our.stdout
#$ -e our.stderr
#$ -q llicen.q
#$ -N OurJob
#$ -cwd
export PATH=/cursos/BI/bin:$PATH # GeneWise, fastaseqfromGFF.pl, t_coffee
##### Para reducir el número de falsos positivos hacemos un nuevo fastasubseq previo al
#SECISearch. El objetivo es que la búsqueda de los elementos SECIS se haga en una región de 10000bp a partir
#de la posición final del hit del tblastn. Para ello tenemos en cuenta la orientación del gen. Para predecir
#elementos SECIS de forma más rigurosa tendríamos que usar un profile y un threshold energético adecuado. Como
#no sabemos que parámetros usar, nuestra predicción de elementos SECIS no es muy buena.
n=$(wc -l ~/perlscripts/scripts_output/bestselenoprotstcoffee | cut -d ' ' -f1);
for line in $(seq 1 $n); do
id=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f2 | tr -d ' ');
chr=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f3 | tr -d ' ');
ini=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f4 | tr -d ' ');
str=$(egrep $id ~/perlscripts/scripts_output/candidates | egrep $ini | cut -f7);
fin=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f5 | tr -d ' ');
if [[ $str != *trev ]]; then
fastasubseq ~/gallopavodb/chromosomes/$chr.fa $fin 10000 > ~/gallopavodb/regions/$id.$chr.SECIS.fa;
fi;
if [[ $str == *trev ]]; then
start=$(($ini - 10000));
fastasubseq ~/gallopavodb/chromosomes/$chr.fa $start 10000 > ~/gallopavodb/regions/$id.$chr.SECIS.fa;
fi;
SECISearch.pl < ~/gallopavodb/regions/$id.$chr.SECIS.fa > ~/our_results/SECIS/$id.$chr.SECIS;
done;

......................................................................................................................................................................................................................................................