
SELENOPROTEÍNAS DE Meleagris gallopavo
MATERIALES Y MÉTODOS: MÓDULO V
MAPA CONCEPTUAL
|
|||
|

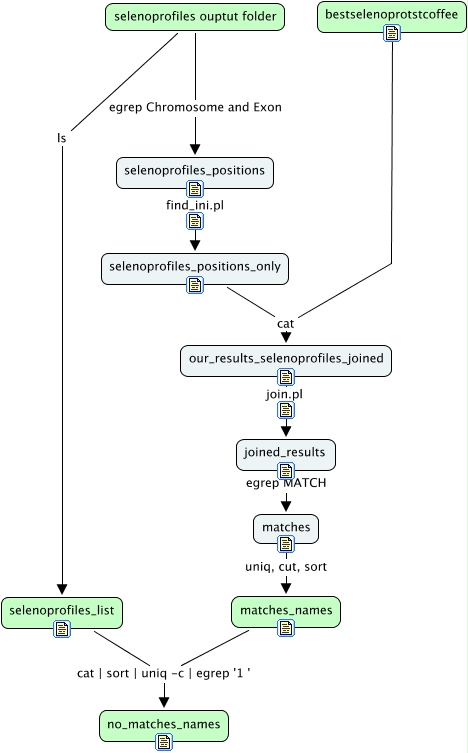
En este módulo comparamos nuestros resultados con los resultados de SelenoProfiles. Hacemos una lista con los resultados contenidos en la carpeta output. Nuevamente nos basamos en la posición en el genoma para encontrar las coincidencias. Esto nos permitió identificar un caso en el que SelenoProfiles había predicho una misma región del genoma como dos selenoproteínas distintas. Comparamos la lista de coincidencias con la lista de todos los archivos de la carpeta output de selenoprofiles y pudimos identificar cuatro selenoproteínas que selenoprofiles había predicho y nosotros no.
BASH script
################################################################################################
################################### - MÓDULO 5 - #####################################
###############################################################################################
###################### Comparación resultados con Selenoprofiles #####################
###############################################################################################
#lista de proteínas predichas por SelenoProfiles
ls ~/results_folder/Meleagris_gallopavo.gallopavo/output/ | egrep *.p2g > ~/perlscripts/scripts_output/selenoprofiles_list;
## generamos un archivo que sólo contiene el nombre de cada archivo, el cromosoma
#y las posiciones de los exones de cada selenoproteína.
n=$(wc -l ~/perlscripts/scripts_output/selenoprofiles_list | cut -d' ' -f1);
for line in $(seq 1 $n); do
file=$(head -$line ~/perlscripts/scripts_output/selenoprofiles_list | tail -1);
echo "#### $file ####" >> ~/perlscripts/scripts_output/selenoprofiles_positions;
chr=$(cat ~/results_folder/Meleagris_gallopavo.gallopavo/output/$file | egrep Chromosome);
position=$(cat ~/results_folder/Meleagris_gallopavo.gallopavo/output/$file | egrep Exon);
echo "$chr" >> ~/perlscripts/scripts_output/selenoprofiles_positions;
echo "$position" >> ~/perlscripts/scripts_output/selenoprofiles_positions;
echo "#### END OF $file ####" >> ~/perlscripts/scripts_output/selenoprofiles_positions;
done;
#<#Con el programa find.ini.pl reducimos el archivo anterior a un archivo que sólo
#contiente el nombre del archivo, el cromosoma y la posición de inicio.
~/perlscripts/find_ini.pl < ~/perlscripts/scripts_output/selenoprofiles_positions > ~/perlscripts/scripts_output/selenoprofiles_positions_only
##concatenamos la lista de nuestros resultados con la de los resultados de selenoprofiles
#y los ordenamos según la posición en el genoma.
cat ~/perlscripts/scripts_output/selenoprofiles_positions_only . ~/perlscripts/scripts_output/bestselenoprotstcoffee | sort -n -k3 -n -k4 >~/perlscripts/scripts_output/our_results_selenoprofiles_joined;
##identificamos los resultados coincidentes basándonos en su posición en el genoma
~/perlscripts/join.pl < ~/perlscripts/scripts_output/our_results_selenoprofiles_joined > ~/perlscripts/scripts_output/joined_results;
egrep 'MATCH' ~/perlscripts/scripts_output/joined_results > ~/perlscripts/scripts_output/matches;
sort ~/perlscripts/scripts_output/matches | uniq | cut -f2 | cut -d' ' -f2 | sort > ~/perlscripts/scripts_output/matches_names;
##identificamos los resultados de selenoprofiles que nosotros no hemos identificado
cat ~/perlscripts/scripts_output/selenoprofiles_list . ~/perlscripts/scripts_output/matches_names | sort | uniq -c | egrep '1 ' > ~/perlscripts/scripts_output/no_matches_names;
##creamos un multifasta con las mejores predicciones para hacer un tblastn contra el
#genoma de gallopavo en ncbi para ver si estos genes están anotados.
n=$(wc -l ~/perlscripts/scripts_output/bestselenoprotstcoffee | cut -d ' ' -f1);
for line in $(seq 1 $n); do
id=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f2 | tr -d ' ');
chr=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f3 | tr -d ' ');
program=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee |tail -1| cut -f8 | tr -d ' ');
if [ $program = 'exonerate' ] ; then
cat ~/our_results/exonerate/aa/$id.$chr.rexonerate.aa >> ~/our_results/bestselenoprots_multifasta.fa;
fi;
if [ $program != 'exonerate' ] ; then
cat ~/our_results/genewise/aa/$id.$chr.genewise.aa >> ~/our_results/bestselenoprots_multifasta.fa;
fi;
done;

......................................................................................................................................................................................................................................................