
SELENOPROTEÍNAS DE Meleagris gallopavo
MATERIALES Y MÉTODOS: MÓDULO III
MAPA CONCEPTUAL
|
|||
|

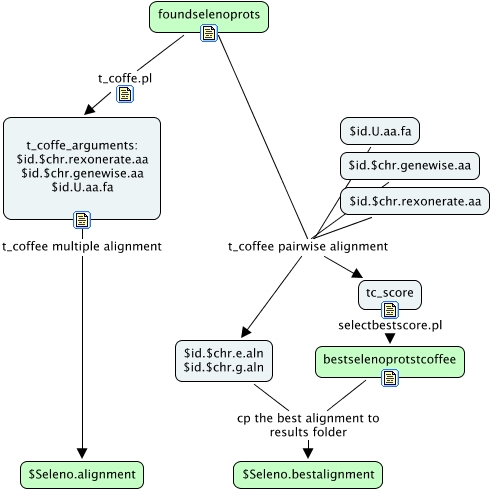
En este módulo hacemos un alineamiento múltiple de todas las proteínas de selenodb que han dado hit en una misma región del genoma. También hacemos pairwise alignments de las predicciones de genewise y exonerate con su query para intentar escoger cual es la proteína más parecida y que software de predicción da un mejor resultado. Esta decisión se basa en el SCORE del pairwise alignment y ya comentaremos en la discusión que no siempre da buenos resultados. Otra opción sería escoger la predicción más larga pero en este caso pierdes información sobre el grado de homología de las distintas proteínas.
BASH script
################################################################################################
################################## - MÓDULO 3 - ######################################
###############################################################################################
############################### FILTRACIÓN RESULTADOS ################################
###############################################################################################
#!/bin/bash
#$ -o our.stdout
#$ -e our.stderr
#$ -q llicen.q
#$ -N OurJob
#$ -cwd
export PATH=/cursos/BI/bin:$PATH
# ### Organización de los resultados: Las secuencias de aminoácidos predichas por EXONERATE
#y GENEWISE se copian a distintas carpetas según su localización en el genoma de 'Meleagris gallopavo'.
#También se copian las proteínas de SELENODB que han dado hit en dicha región.
n=$(wc -l ~/perlscripts/scripts_output/foundselenoprots | cut -d ' ' -f1);
for line in $(seq 1 $n); do
id=$(head -$line ~/perlscripts/scripts_output/foundselenoprots | tail -1 | cut -f5 | tr -d ' ');
chr=$(head -$line ~/perlscripts/scripts_output/foundselenoprots | tail -1 | cut -f2 | tr -d ' ');
prot=$(head -$line ~/perlscripts/scripts_output/foundselenoprots | tail -1 | cut -f1 | tr -d ' ');
mkdir ~/our_results/gallopavo_selenoproteins/$prot;
cp ~/our_results/genewise/aa/$id.$chr.genewise.aa ~/our_results/gallopavo_selenoproteins/$prot;
cp ~/our_results/exonerate/aa/$id.$chr.rexonerate.aa ~/our_results/gallopavo_selenoproteins/$prot;
cp ~/gallopavodb/selenoprots/Uselenoprots/$id.U.aa.fa; ~/our_results/gallopavo_selenoproteins/$prot;
done;
# ############################## T_COFFEE MULTIPLE-ALIGNMENT ###############################
# Se realiza un 'multiple-alignment' con t_coffee de todos los hits para cada región (SelenoX) y también incluyendo
# la 'QUERY' DE SELENODB
cat foundselenoprots . END > foundselenoprotsEND;
# # "foundselenoprotsEND": Se añade línea al final para el correcto funcionamiento de t_coffee.pl
#t_coffee.pl > Genera un archivo que contiene los argumentos necesarios para hacer los alineamientos múltiples con
#t_coffee llamado "t_coffee_arguments"
~/perlscripts/t_coffee.pl < ~/perlscripts/scripts_output/foundselenoprotsEND > ~/perlscripts/scripts_output/t_coffee_arguments;
n=$(wc -l ~/perlscripts/scripts_output/t_coffee_arguments | cut -d ' ' -f1);
for line in $(seq 1 $n); do
prot=$(head -$line ~/perlscripts/scripts_output/t_coffee_arguments | tail -1 | cut -f1 | tr -d ' ');
arguments=$(head -$line ~/perlscripts/scripts_output/t_coffee_arguments | tail -1 | cut -f2);
t_coffee $arguments > ~/our_results/gallopavo_selenoproteins/$prot/$prot.alignment;
done;
################################## T_COFFEE PAIRWISE-ALIGNMENT ############################
# Con tal de simplificar los resultados y saber la família de selenoproteína a la que pertenece cada proteína decidimos
#utilizar el SCORE de pairwise alignments de t_coffee. Veremos que esta decisión le da mucha importancia a la homología
#pero muy poca a la longitud de la predicción. Esto tiene sus ventajas y sus inconvenientes.
n=$(wc -l ~/perlscripts/scripts_output/foundselenoprots | cut -d ' ' -f1);
for line in $(seq 1 $n); do
row=$(head -$line ~/perlscripts/scripts_output/foundselenoprots | tail -1);
id=$(head -$line ~/perlscripts/scripts_output/foundselenoprots | tail -1 | cut -f5 | tr -d ' ');
chr=$(head -$line ~/perlscripts/scripts_output/foundselenoprots | tail -1 | cut -f2 | tr -d ' ');
## Pairwise-alignment (exonerate)
t_coffee ~/gallopavodb/selenoprots/Uselenoprots/$id.U.aa.fa ~/our_results/exonerate/aa/$id.$chr.rexonerate.aa -outfile=~/perlscripts/scripts_output/t_coffee/$id.$chr.e.aln -quiet;
scoreex=$(egrep SCORE ~/perlscripts/scripts_output/t_coffee/$id.$chr.e.aln | cut -d'=' -f3 | cut -d',' -f1);
## Pairwise-alignment (genewise)
t_coffee ~/gallopavodb/selenoprots/Uselenoprots/$id.U.aa.fa ~/our_results/genewise/aa/$id.$chr.genewise.aa -outfile=~/perlscripts/scripts_output/t_coffee/$id.$chr.g.aln -quiet;
scoregen=$(egrep SCORE ~/perlscripts/scripts_output/t_coffee/$id.$chr.g.aln | cut -d'=' -f3 | cut -d',' -f1);
# # Elección de la mejor predicción (Exonerate o Genewise) para cada proteína según el SCORE
#del t_coffee. En el archivo tc_score se muestra la puntuación y el programa elegido. Si el score de exonerate y genewise
#son iguales, por defecto se muestra el alineamiento de genewise pero esta decisión es arbitraria.
if [ $scoreex -gt $scoregen ] ; then
echo -e "$row \t $scoreex \t exonerate" >> ~/perlscripts/scripts_output/t_coffee/tc_score;
fi;
if [ $scoregen -gt $scoreex ] ; then
echo -e "$row \t $scoregen \t genewise" >> ~/perlscripts/scripts_output/t_coffee/tc_score;
fi;
if [ $scoreex = $scoregen ] ; then
echo -e "$row \t $scoreex \t exonerate/genewise" >> ~/perlscripts/scripts_output/t_coffee/tc_score;
fi;
scoreex=$(' ');
scoregen=$(' ');
done;
# #### ELECCIÓN DEL MEJOR ALINEAMIENTO PARA CADA SELENOPROTEÍNA PREDICHA ####
#Nos basamos en que, a pesar de que varias proteínas de selenodb tengan homología por una misma región del genoma
#de M. gallopavo, todas ellas nos informan de la existencia de una sola selenoproteína. Con selectbestscore.pl escogemos
#la selenoproteína más parecida a la nuestra, nuevamente basándonos en el SCORE de los pairwise alignments. Generamos el
#archivo bestselenoprotstcoffee. Esta decisión es importante para definir la família a la que podría pertenecer la proteína
#predicha. Como hemos comentado, la elección de la mejor predicción no siempre ha funcionado bien. Dado que todas las
#proteínas de cada multiple alignment son de la misma família, esta información no se ve seriamente afectada por la elección
#del mejor alineamiento.
cat ~/perlscripts/scripts_output/t_coffee/tc_score . ~/perlscripts/scripts_output/END > ~/perlscripts/scripts_output/tc_scoreEND;
~/perlscripts/selectbestscore.pl < ~/perlscripts/scripts_output/tc_scoreEND > ~/perlscripts/scripts_output/bestselenoprotstcoffee;
# #### ALMACENAMIENTO DEL MEJOR ALINEAMIENTO DEL PAIRWISE ALIGNMENT EN LAS CARPETAS DE LAS SELENOPROTEÍNAS ENCONTRADAS####
n=$(wc -l ~/perlscripts/scripts_output/bestselenoprotstcoffee | cut -d ' ' -f1);
for line in $(seq 1 $n); do
prot=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f1 | tr -d ' ');
id=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f2 | tr -d ' ');
chr=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee | tail -1 | cut -f3 | tr -d ' ');
program=$(head -$line ~/perlscripts/scripts_output/bestselenoprotstcoffee |tail -1| cut -f8 | tr -d ' ');
if [ $program = 'exonerate' ] ; then
cp ~/perlscripts/scripts_output/t_coffee/$id.$chr.e.aln ~/our_results/gallopavo_selenoproteins/$prot/$prot.bestalignment;
fi;
if [ $program != 'exonerate' ] ; then
cp ~/perlscripts/scripts_output/t_coffee/$id.$chr.g.aln ~/our_results/gallopavo_selenoproteins/$prot/$prot.bestalignment;
fi;
done;

......................................................................................................................................................................................................................................................