La proteína UFD2/D4COLE1E se forma por splicing conjunto, a través de los límites del dominio de triplicación, de exones de los genes Nmnat1 y Ube4b, codificantes de las proteínas D4COLE1E y UFD2, respectivamente. UFD2 codifica para un factor de poliubiquitinización; mientras que, D4COLE1E codifica para un enzima sintetizador de NAD+.
El dominio N-terminal de ésta proteína de fusión está formado por 70 aminoácidos del dominio esencial para la ubiquitinización de UFD2. Esta región viene seguida por un ácido aspártico conector y la secuencia codificante, al completo, de Nmnat1(302 aminoácidos).
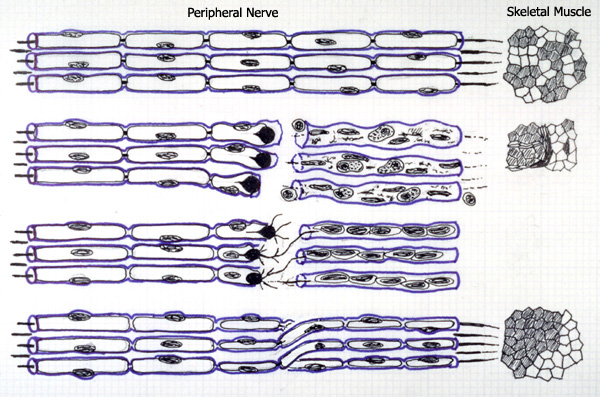
Estudios recientes hipotetizan que esta proteína de fusión estaría involucrada en el retardo del proceso conocido como degeneración de Wallerian, por el cual la parte distal de un axón perteneciente a una fibra rota/dañada, degenera. La relevancia de este proceso depende de la localización, central o periférica, del daño.
Los restos de axones y de la capa de mielina son digeridos por macrófagos y células de Schwann, éstas solo están presentes en el sistema nervioso periférico. Como consecuencia de la falta de inervación, el tejido diana (en SNP, normalmente un músculo) se atrofia.
Se puede dar la regeneración de la parte terminal del axón gracias a factores de crecimiento segregados por las células de Schwann cercanas a la lesión y también, debido al rebrote axonal. A menudo, si la lesión es muy grande o se ha formado tejido cicatrizal, dicha regeneración se ve dificultada o relentizada. En el sistema nervioso central, la falta de células de Schwann dificulta el proceso, siendo casi exclusivo del SNP.
Estudios hechos con ratones mutantes(WldS), que expresan la proteína de fusión y, por lo tanto, una degeneración retardada, han permitido determinar los distintos tipos celulares involucrados, así como, los procesos moleculares subyacentes.